Stała szybkości reakcji chemicznej. Stała szybkości reakcji Wytyczne dotyczące prac laboratoryjnych
Zgodnie z prawem działania mas, szybkość prostej reakcji jest równa
Stała szybkości reakcji k
-
współczynnik proporcjonalności między szybkością reakcji chemicznej a iloczynem stężeń substancji reagujących:  . Stała szybkości jest liczbowo równa szybkości reakcji chemicznej, gdy stężenia wszystkich reagentów są równe jedności: W=k przy C A =C B =1. Jeżeli reakcja A z B jest złożona pod względem mechanizmu (zawiera aktywne produkty pośrednie, katalizator itp.), jest zgodna z równaniem
. Stała szybkości jest liczbowo równa szybkości reakcji chemicznej, gdy stężenia wszystkich reagentów są równe jedności: W=k przy C A =C B =1. Jeżeli reakcja A z B jest złożona pod względem mechanizmu (zawiera aktywne produkty pośrednie, katalizator itp.), jest zgodna z równaniem  , wówczas nazywa się k stała efektywnej szybkości reakcji; IUPAC zaleca w tym przypadku wywołanie k współczynnik szybkości reakcji. Często szybkość reakcji złożonej nie jest zgodna z równaniem potęgowym, lecz wyrażana inną zależnością, np. v=k 1 C 1 C 2 (1+k 2 C 2) –1. Następnie wywoływane są k 1 i k 2 współczynniki w równaniu szybkości reakcji.
, wówczas nazywa się k stała efektywnej szybkości reakcji; IUPAC zaleca w tym przypadku wywołanie k współczynnik szybkości reakcji. Często szybkość reakcji złożonej nie jest zgodna z równaniem potęgowym, lecz wyrażana inną zależnością, np. v=k 1 C 1 C 2 (1+k 2 C 2) –1. Następnie wywoływane są k 1 i k 2 współczynniki w równaniu szybkości reakcji.
Często reakcję prowadzi się w warunkach, w których stężenia wszystkich odczynników, z wyjątkiem jednego, są przyjmowane w nadmiarze i praktycznie nie zmieniają się w trakcie eksperymentu. W tym przypadku
 ,
,
i współczynnik k obs. =
k  zwany skuteczny Lub obserwowana stała szybkości reakcji w C B >> C A . Dla przypadku n A = 1 taki współczynnik nazywany jest często współczynnikiem szybkości reakcji pseudo pierwszego rzędu. Stała szybkości reakcji rzędu n ma wymiar: (czas) –1 (stężenie) –(n –1) . Wartość liczbowa zależy od jednostek wybranych do pomiaru czasu i stężenia.
zwany skuteczny Lub obserwowana stała szybkości reakcji w C B >> C A . Dla przypadku n A = 1 taki współczynnik nazywany jest często współczynnikiem szybkości reakcji pseudo pierwszego rzędu. Stała szybkości reakcji rzędu n ma wymiar: (czas) –1 (stężenie) –(n –1) . Wartość liczbowa zależy od jednostek wybranych do pomiaru czasu i stężenia.
Obliczając stałą szybkości prostej reakcji należy wziąć pod uwagę dwie okoliczności: pamiętać od jakiego odczynnika mierzona jest szybkość reakcji oraz jaki jest współczynnik stechiometryczny i kolejność reakcji dla tego odczynnika. Na przykład reakcja rodnika 2,4,6-trialkilofenoksylowego z wodoronadtlenkiem zachodzi w dwóch kolejnych etapach:
PhО +ROOH → PhOH+RO 2
PhO +RO 2 →ROOPhO
Równanie stechiometryczne to 2PhO +ROOH=PhOH+ROOPhO, ale ponieważ pierwszy etap określa szybkość reakcji, W ROOH =k i W PhO =2k.
Zatem współczynniki w równaniach kinetycznych i stechiometrycznych dla rodnika fenoksylowego nie pokrywają się tutaj: rząd reakcji w PhO wynosi 1, a współczynnik stechiometryczny dla PhO wynosi 2.
Metody obliczania stałej szybkości reakcji chemicznej. Zgodnie z krzywą kinetyczną. Jeśli n = 1, wtedy k=t –1 ln 10 lg (C Ao /C A). Jeśli całkowity rząd reakcji wynosi n, a rząd reakcji dla danego składnika wynosi 1, a wszystkie odczynniki oprócz A zostały pobrane w nadmiarze, to
 .
.
Dla reakcji A+B → produkty k znajdują się z równania

Przy obliczaniu stałej szybkości z całkowej krzywej kinetycznej w postaci ogólnej zadaniem jest wyznaczenie k w równaniu f(x)= –k`t (x jest względnym stężeniem odczynnika).
Dla reakcji pierwszego rzędu f(x)=ln x, k`=k; dla reakcji II rzędu f(x)=x –1 –1, k=C o k, itd. Z eksperymentu otrzymujemy szereg wartości (t 1, x 1), (t 2, x 2), ..., (t n, x n). Linia prosta poprowadzona we współrzędnych f(x)–t musi spełniać warunek i =f(x i)+kt i, Σ i =0. Wynika z tego, że k= Σf(x i)/Σt i.
Według okresu półtrwania. Okres półtrwania jest jednoznacznie powiązany ze stałą szybkości i początkowym stężeniem odczynnika, co pozwala nam obliczyć k. Zatem dla reakcji pierwszego rzędu k=ln 2/τ 1/2, dla reakcji drugiego rzędu k=C o –1 τ 1/2 itd.
Zgodnie z początkową szybkością reakcji. Ponieważ w początkowej chwili zużycie odczynników jest niewielkie,
 I
I 
Przez zmianę szybkości reakcji w czasie. Mierząc stężenia reagentów w czasie t` i t`` (С` i С``), możemy obliczyć średnią szybkość reakcji i znaleźć k, przy ν=1 mamy
 ,
,
 ,
,
 .
.
Specjalne metody przetwarzania krzywych kinetycznych. Jeżeli kinetykę reakcji rejestruje się poprzez zmianę dowolnej właściwości fizycznej układu x (gęstość optyczna, przewodność elektryczna itp.) związanej ze stężeniem reagenta C tak, że przy C=C o , x=x o i przy C=0 , x=x ∞ wówczas k można wyznaczyć z krzywej kinetycznej x(t) stosując następujące metody:
Metoda Guggenheima(dla reakcji pierwszego rzędu). Zmierz x i w czasie t i i x 1 ` w momencie t i + itd. Z wykresu lg (х i –х i`)–t znajduję k:
log (x i –x i `)=log[(x o –x ∞)(1–e – k )]–0,43kt i .
Metoda Mangelsdorffa(dla reakcji pierwszego rzędu). Pomiary przeprowadza się jak w metodzie Guggenheima, z tym że wykres wykreślany jest we współrzędnych x i ` – x i:
x i `=x i e –k +x ∞ (1–e –k ),
nachylenie prostej wynosi e – k , punkt przecięcia na osi rzędnych jest równy x ∞ (1 – e – k ).
Metoda Rosevery’ego(dla reakcji drugiego rzędu). Parametr x mierzony jest w momentach t 1, t 2, t 3 oddzielonych stałym odstępem czasu . Stałą szybkości oblicza się z równania:
 .
.
Rozdział 5. KINETYKA REAKCJI CHEMICZNYCH I KATALIZA.
Reakcje możliwe termodynamicznie nie zawsze są przeprowadzane w rzeczywistości. Wynika to z faktu, że w termodynamice nie ma parametru czasu, więc nie odpowiada on, jak szybko nastąpi dany stan. Określenie warunków, w których możliwe termodynamicznie reakcje będą przebiegać z wystarczającą szybkością, jest jednym z głównych problemów kinetyki chemicznej. W kinetyce wprowadza się czynnik czasu, który nie jest uwzględniany w termodynamice.
Kinetyka chemiczna - jest to doktryna o regularności przebiegu procesu chemicznego w czasie lub doktryna o mechanizmach i szybkości reakcji chemicznych.
Zbiór etapów składających się na reakcję chemiczną nazywa się mechanizm lub schemat reakcji chemicznej.
Szybkość reakcji chemicznej.
Pod szybkością reakcji chemicznej zrozumieć zmianę liczby moli reagujących substancji w jednostce czasu na jednostkę objętości.
Istnieją średnie prędkości ( ty por) i prawda ( ty).
Średnia prędkość - zmiana stężenia reagujących substancji w zadanym okresie czasu:
u av = ± (n 2 – n 1) / V(t 2 - t 1) = ± Dn / V Δt = ± Δс / Δt.
Stosunek Δс/Δt może być dodatni lub ujemny. Prędkość można zmierzyć monitorując spadek stężenia związku wyjściowego, wówczas przed stosunkiem stawiamy znak minus, ponieważ prędkość ma zawsze wartość dodatnią. Jeżeli prędkość wyraża się poprzez stężenie substancji odbierającej, wówczas znak plus:
- Δс А / Δt= + Δс В /Δt.
Możesz powiązać zmianę stężenia z nieskończenie małym okresem czasu (t 2 -t 1 → 0), definiując rzeczywista szybkość reakcji w danej chwili jako pochodna stężenia po czasie (u = ±dс/dt).
- dc A /dt = + dc B /dt
Zależność szybkości reakcji od stężenia.
Głównym postulatem kinetyki chemicznej jest prawo działania mas ustalone przez Guldberga i Vahe. Rozważ reakcję chemiczną:
m 1 A + m 2 B → m 3 C + m 4 D.
Nazywa się równanie opisujące zależność szybkości reakcji chemicznej od stężenia składników mieszaniny reakcyjnej równanie kinetyczne reakcji chemicznej.
Równanie kinetyczne rozważanej reakcji:
u = kс A m 1 `s B m 2 ,
gdzie k jest współczynnikiem proporcjonalności (stała prędkości).
Prawo działania mas: szybkość reakcji chemicznej w każdym momencie jest wprost proporcjonalna do iloczynu stężeń substancji reagujących w danym momencie w potęgach odpowiadających stechiometrycznym współczynnikom reakcji (w najprostszym przypadku).
W większości przypadków nie jest obliczana prędkość, ale stała prędkość. Jeśli c A = c B = 1 mol/l, to ty = k.
Fizyczne znaczenie stałej szybkości: stała szybkości reakcji chemicznej jest liczbowo równa szybkości reakcji, pod warunkiem, że stężenia reagentów są stałe i równe jedności. Stała szybkości jest niezależna od stężenia i zależy od temperatury, rodzaju rozpuszczalnika i obecności katalizatora.
Wszystkie reakcje są kinetycznie dwukierunkowe lub kinetycznie odwracalne. Reakcja chemiczna jest odwracalna, gdy produkty reakcji mogą ze sobą reagować, tworząc materiały wyjściowe. W praktyce reakcja odwrotna może być tak powolna w porównaniu do reakcji bezpośredniej, że przy jakiejkolwiek rozsądnej dokładności można pominąć odwracalność reakcji i uznać reakcję za nieodwracalną lub jednostronną. Ściśle mówiąc, wszelkie reakcje chemiczne są odwracalne:
m 1 A 4 +m 2 B « m 3 C+m 4 D
u = u 1 - u 2 = k 1 s A m 1 `s B m 2 - k 2 s Z m 3 D m 4,
W momencie równowagi chemicznej u 1 = u 2 , te
k 1 s A m 1 `s B m 2 = k 2 s Z m 3 D m 4,
DO =k 1 / k 2=Z Z m 3´ Z D m 4/ Z A m 1´ Z B m 2
gdzie K jest stałą równowagi chemicznej, równą stosunkowi stałej szybkości reakcji naprzód do stałej szybkości reakcji odwrotnej.
Klasyfikacja reakcji ze względu na molekularność i porządek.
Podczas badania kinetyki reakcje chemiczne różnią się molekularnością i porządkiem.
Molekularność reakcji zależy od liczby cząsteczek uczestniczących jednocześnie w etapie, który określa szybkość całej reakcji (najwolniej). Na tej podstawie reakcje dzieli się na mono-, bi- i trimolekularne. Reakcje o wyższej masie cząsteczkowej są praktycznie nieznane, ponieważ prawdopodobieństwo spotkania czterech cząsteczek jest znikome.
Kolejność reakcji jest określana przez sumę wykładników stężeń w wyrażeniu prawa działania mas. Rozróżnia się kolejność reakcji pełną (ogólną) i częściową (dla każdego odczynnika). Suma wykładników, w których stężenia wszystkich substancji wyjściowych wchodzą do równania kinetycznego, określa ogólny porządek. Istnieją reakcje rzędu zerowego, pierwszego, drugiego, trzeciego i ułamkowego.
Zbieżność molekularności z porządkiem obserwuje się tylko w najprostszych przypadkach, gdy reakcja przebiega jednoetapowo:
2NO + H 2 ↔ N 2 O + H 2 O,
porządek ogólny - 3, molekularność - 3.
5.3.1. Równanie reakcji jednokierunkowej pierwszego rzędu.
Rozważmy reakcję chemiczną: A → B.
u = kс = - dс/dt.
Rozdzielmy zmienne: -dс/с = k dt, całkuj
Lnc = kt + stała,
jeśli τ = 0 (początkowy moment reakcji), to const = ln c 0, tj.
Ln с = kt - ln с 0,
ln s 0 - ln s = kt lub ln s 0 / s = kt,
k = (1/t)’ ln s 0 /s.
Oznaczmy x - stopień przemiany substancji wyjściowej: x = c 0 – c.
k = (1/t) ´ln s 0 /(s 0 - x),
wymiar - [czas -1 ].
Stała szybkości reakcji pierwszego rzędu jest niezależna od stężenia. Do otrzymanego równania można zastąpić stężenie (mol/l) lub liczbę moli. Zamiast „c 0” i „(c 0 - x)” można zastąpić dowolne wartości proporcjonalne do stężenia (przewodność elektryczna, gęstość, lepkość itp.).
Aby scharakteryzować szybkość reakcji pierwszego rzędu, wraz ze stałą szybkości, często stosuje się wielkość zwaną okresem półtrwania.
Pół życia(t 1/2)- okres czasu, w którym połowa przyjętej ilości substancji reaguje:
t 1/2 = (1/k)’ ln s 0 /(s 0 - x), gdzie x = 1/2s 0.
Otrzymujemy:
t 1/2 = ln2/k = 0,693/k.
Okres półtrwania nie zależy od stężeń początkowych, ale zależy od stałej szybkości, tj. jest to cecha reakcji pierwszego rzędu.
Reakcje pierwszego rzędu obejmują rozpad radioaktywny, izomeryzację i większość reakcji hydrolizy. Gdy występuje duży nadmiar jednego z reagentów w stosunku do pozostałych, jego stężenie podczas reakcji pozostaje prawie stałe. W tym przypadku rząd reakcji będzie o jeden mniejszy niż można by oczekiwać na podstawie równania stechiometrycznego.
Reakcje dwucząsteczkowe, w których rząd reakcji pod wpływem nadmiaru jednego z reagentów zmniejsza się o jeden, nazywane są pseudocząsteczkowymi.
Przykład reakcji hydrolitycznego rozkładu cukru w rozcieńczonym roztworze wodnym (inwersja cukru):
C 12 H 22 O 11 + H 2 O ↔ C 12 H 22 O 11 + C 12 H 22 O 11
sacharoza glukoza fruktoza
u = k[sacharoza]’,
u = k* [sacharoza], gdzie k* = k´.
To jest przykład reakcji pseudo pierwszego rzędu.
Równanie reakcji jednokierunkowej drugiego rzędu.
A + B → C + D
Przykład: H2 + J2 = 2HJ;
2HJ = H2 + J2;
CH 3 COOC 2 H 5 + NaOH = CH 3 COONA + C 2 H 5 OH.
Dс/dt = ks 1 `с 2
Gdy с 1 = с 2 otrzymujemy: -dс/dt =kс 2 lub -dс/с 2 = k dt. Zintegrujmy:
1/s = kt + stała
Przy t = 0 → const = 1/s 0.
1/s - 1/s 0 = kt lub (s 0 – s)/s's 0 = kt;
c 0 - c = x, gdzie x to stopień konwersji; do = do 0 - x;
x / s 0 (s 0 - s) = kt;
k = (1/ t)’,
wymiar - [czas -1 `stężenie -1 ).
Stała szybkości reakcji drugiego rzędu zależy od wymiaru stężenia.
Okres półtrwania: t 1/2 = (1/ k), gdzie x = 1/2s 0, wówczas
t 1/2 = 1/ ks 0 .
Okres półtrwania zależy od stężenia początkowego i nie jest cechą charakterystyczną reakcji drugiego rzędu.
Równanie reakcji zerowego rzędu.
Szybkość reakcji chemicznej nie zależy od stężenia reagentów (reakcje na granicy faz, procesem ograniczającym jest proces dyfuzji):
Dc/dt = ks 0 ; lub -dс = k dt.
Całkujemy i otrzymujemy: -с = kt + const.
Przy t = 0 → const = -с 0. Otrzymujemy: -с = kt - с 0;
k = (c 0 - c) /t = x/t,
wymiar - [stężenie czas -1].
Pół życia:
t 1/2 = do 0 /2k
Metody wyznaczania rzędu i stałej szybkości reakcji.
W kinetyce reakcji typów prostych i złożonych rozwiązuje się głównie następujące problemy:
1. Zadanie bezpośrednie: znany jest porządek reakcji i stała jej szybkości. Należy znaleźć stężenie którejkolwiek z substancji wyjściowych lub produktów reakcji w określonym momencie lub znaleźć czas, w którym stężenie któregokolwiek z reagentów lub produktów reakcji osiąga określoną wartość.
2. Problem odwrotny: uzyskano dane eksperymentalne dotyczące kinetyki wcześniej niezbadanej reakcji. Wymagane jest określenie rzędu i stałej szybkości reakcji.
Aby określić kolejność reakcji, konieczne jest posiadanie danych eksperymentalnych dotyczących zmian stężenia reagentów w czasie:
| od 0 | od 1 | od 2 | od 3 | od 4 | ….. |
| t 0 | t 1 | t 2 | t 3 | t 4 | ….. |
1. Metoda doboru równań.
Metoda polega na podstawieniu danych eksperymentalnych dotyczących stężeń substancji w każdym momencie od początku reakcji do równań kinetycznych różnych rzędów (technika ta nic nie daje, jeśli rząd reakcji jest większy niż 3 lub jest ułamkowy):
k = (s 0 - s) /t = x/t(rząd zerowy);
k = (1/t) lnc 0 /s(Pierwsze zamówienie);
k = (1/t) x /s 0 s ( drugie zamówienie).
Kolejność reakcji będzie odpowiadać równaniu kinetycznemu, dla którego przy różnych początkowych stężeniach substancji wyjściowych i w różnych czasach w danej temperaturze stała szybkości będzie wartością stałą.
2. Graficzne metody całkowe.
 |  |
rząd zerowy: rząd pierwszy, drugi
Ryż. 5.1. Stężenie zmienia się w czasie reakcji
różne zamówienia.
Znajdź taką funkcję stężenia, nanosząc ją na wykres w zależności od czasu, aby uzyskać linię prostą (rys. 5.1.).
3. Przez okres półtrwania.
Zgodnie z zależnością okresu półtrwania od stężenia początkowego:
rząd zerowy: t 1/2 = c 0 /2k;
pierwszego rzędu: t 1/2 = 0,693/k;
drugiego rzędu: t 1/2 = 1 / ks 0.
Ogólnie:
t 1/2 ≈ 1 /k s 0 n-1.
Doświadczenia przeprowadza się przy dwóch różnych stężeniach początkowych (od 0)” i (od 0)”:
(t 1/2) ’ = 1 /k (s 0) 1 n-1 (1)
(t 1/2)” = 1 /k (s 0) 2 n-1 (2)
Podzielmy (1) przez (2):
(t 1/2) ’ / (t 1/2)” = (s 0) 2 n-1 / (s 0) 1 n-1 .
Weźmy logarytm:
log(t 1/2) ’ / (t 1/2)” = (n-1) ´ log[(s 0) 2 /(s 0) 1 ],
n = 1 + / .
4.Metoda różniczkowa (metoda van't Hoffa).
Wykorzystuje się zależność szybkości reakcji od stężenia, pod warunkiem, że stężenia wszystkich substancji wyjściowych są równe (ryc. 5.2.): u = kс n. Weźmy logarytm tego wyrażenia: lgu = lgk + nlgс.

Ryż. 5.2. Zależność szybkości reakcji od stężenia.
5. Całkowa metoda van't Hoffa (w zależności od szybkości reakcji od początkowego stężenia w pierwszych chwilach czasu - 10-15 s).
u = k (do 0 - x) n = k do 0 n ,
Ponieważ w pierwszym momencie czasu x ≈ 0.
Doświadczenie przeprowadza się przy różnych stężeniach początkowych.
u 1 = k do 1 n (1)
u 2 = k do 2 n (2)
Równanie (1) dzielimy przez równanie (2): u 1 / u 2 = (c 1 / c 2) n.
Weźmy logarytmy:
n = (lgu 1 - lgu 2) / (lgс 1 -lgс 2),
gdzie c 1 i c 2 przyjmuje się jako średnie dla badanego odcinka reakcji, odpowiadające Δt.
6. Metoda izolacji Ostwalda.
Zapiszmy równanie kinetyczne reakcji: u = kс A n 1 `с B n 2 `с Z n 3.
Zwiększamy stężenie „B” i „C” ponad 10-krotnie. Kolejność tych substancji będzie wynosić zero, ich stężenia nie ulegną zmianie. „n 1” wyznaczamy jedną z metod omówionych powyżej. To samo robimy ustalając rząd reakcji w oparciu o substancje B i C, tj. n 2 i n 3.
1. Podstawowe pojęcia i postulaty kinetyki chemicznej
Kinetyka chemiczna to dział chemii fizycznej zajmujący się badaniem szybkości reakcji chemicznych. Główne zadania kinetyki chemicznej: 1) obliczanie szybkości reakcji i wyznaczanie krzywych kinetycznych, tj. zależność stężeń reagentów od czasu ( bezpośrednie zadanie); 2) wyznaczanie mechanizmów reakcji z krzywych kinetycznych ( problem odwrotny).
Szybkość reakcji chemicznej opisuje zmianę stężeń reagentów w jednostce czasu. Dla reakcji
A+ B B+... D D+ mi E+...
szybkość reakcji określa się w następujący sposób:
gdzie nawiasy kwadratowe wskazują stężenie substancji (zwykle mierzone w mol/l), T- czas; A, B, D, mi- współczynniki stechiometryczne w równaniu reakcji.
Szybkość reakcji zależy od rodzaju reagentów, ich stężenia, temperatury i obecności katalizatora. Zależność szybkości reakcji od stężenia opisuje podstawowy postulat kinetyki chemicznej - prawo akcji masowej:
Szybkość reakcji chemicznej w każdym momencie jest proporcjonalna do aktualnego stężenia reagentów podniesionego do pewnych potęg:
![]() ,
,
Gdzie k- stała szybkości (niezależna od stężenia); X, y- niektóre numery, które są wywoływane kolejność reakcji według substancji Odpowiednio A i B. Ogólnie rzecz biorąc, liczby te nie mają nic wspólnego ze współczynnikami A I B w równaniu reakcji. Suma wykładników X+ y zwany ogólny porządek reakcji. Kolejność reakcji może być dodatnia lub ujemna, całkowita lub ułamkowa.
Większość reakcji chemicznych składa się z kilku etapów zwanych reakcje elementarne. Przez reakcję elementarną rozumie się zazwyczaj pojedynczy akt utworzenia lub rozerwania wiązania chemicznego, przebiegający poprzez utworzenie kompleksu przejściowego. Nazywa się liczbą cząstek biorących udział w reakcji elementarnej molekularność reakcje. Istnieją tylko trzy rodzaje reakcji elementarnych: monocząsteczkowe (A B + ...), dwucząsteczkowe (A + B D + ...) i trójcząsteczkowe (2A + B D + ...). W przypadku reakcji elementarnych ogólny porządek jest równy molekularności, a porządki według substancji są równe współczynnikom w równaniu reakcji.
PRZYKŁADY
Przykład 1-1. Szybkość tworzenia NO w reakcji 2NOBr (g) 2NO (g) + Br2 (g) wynosi 1,6. 10-4 mol/(l.s). Jaka jest szybkość reakcji i zużycie NOBr?
Rozwiązanie. Z definicji szybkość reakcji wynosi:
Mol/(l.s).
Z tej samej definicji wynika, że stopień zużycia NOBr jest równy szybkości tworzenia NO o przeciwnym znaku:
![]() mol/(l.s).
mol/(l.s).
Przykład 1-2. W reakcji II rzędu A + B D początkowe stężenia substancji A i B wynoszą odpowiednio 2,0 mol/L i 3,0 mol/L. Szybkość reakcji wynosi 1,2. 10 -3 mol/(l.s) przy [A] = 1,5 mol/l. Oblicz stałą szybkości i szybkość reakcji przy [B] = 1,5 mol/l.
Rozwiązanie. Zgodnie z prawem działania mas, w dowolnym momencie szybkość reakcji jest równa:
![]() .
.
Do czasu, gdy [A] = 1,5 mol/l, przereagowało 0,5 mol/l substancji A i B, zatem [B] = 3 – 0,5 = 2,5 mol/l. Stała szybkości wynosi:
L/(mol. s).
Do czasu, gdy [B] = 1,5 mol/l, przereagowało 1,5 mol/l substancji A i B, zatem [A] = 2 – 1,5 = 0,5 mol/l. Szybkość reakcji wynosi:
Mol/(l.s).
ZADANIA
1-1. Jak szybkość reakcji syntezy amoniaku 1/2 N 2 + 3/2 H 2 = NH 3 wyraża się w postaci stężeń azotu i wodoru? (odpowiedź)
1-2. Jak zmieni się szybkość reakcji syntezy amoniaku 1/2 N 2 + 3/2 H 2 = NH 3, jeśli równanie reakcji zapiszemy jako N 2 + 3H 2 = 2NH 3? (odpowiedź)
1-3. Jaka jest kolejność reakcji elementarnych: a) Cl + H 2 = HCl + H; b) 2NO + Cl2 = 2NOCl? (odpowiedź)
1-4. Która z poniższych wielkości może przyjąć a) ujemną; b) wartości ułamkowe: szybkość reakcji, rząd reakcji, molekularność reakcji, stała szybkości, współczynnik stechiometryczny? (odpowiedź)
1-5. Czy szybkość reakcji zależy od stężenia produktów reakcji? (odpowiedź)
1-6. Ile razy wzrośnie szybkość elementarnej reakcji w fazie gazowej A = 2D, gdy ciśnienie wzrośnie 3-krotnie? (odpowiedź)
1-7. Określ rząd reakcji, jeśli stała szybkości ma wymiar l 2 / (mol 2 . s). (odpowiedź)
1-8. Stała szybkości reakcji gazowej drugiego rzędu w temperaturze 25 o C wynosi 10 3 l/(mol. s). Ile wynosi ta stała, jeśli równanie kinetyczne wyraża się w postaci ciśnienia w atmosferach? (odpowiedź)
1-9. Do reakcji w fazie gazowej N rzędu nA B, wyraź szybkość tworzenia się B w postaci całkowitego ciśnienia. (odpowiedź)
1-10. Stałe szybkości reakcji w przód i w tył wynoszą 2,2 i 3,8 l/(mol. s). Dzięki któremu z poniższych mechanizmów mogą zachodzić te reakcje: a) A + B = D; b) A + B = 2D; c) A = B + D; d) 2A = B.(odpowiedź)
1-11. Reakcja rozkładu 2HI H 2 + I 2 jest drugiego rzędu ze stałą szybkością k= 5,95. 10 -6 l/(mol. s). Oblicz szybkość reakcji pod ciśnieniem 1 atm i temperaturze 600 K. (odpowiedź)
1-12. Szybkość reakcji drugiego rzędu A + B D wynosi 2,7. 10 -7 mol/(l.s) przy stężeniach substancji A i B odpowiednio 3,0. 10 -3 mol/l i 2,0 mol/l. Oblicz stałą szybkości. (odpowiedź)
1-13. W reakcji II rzędu A + B 2D początkowe stężenia substancji A i B wynoszą 1,5 mol/l. Szybkość reakcji wynosi 2,0. 10 -4 mol/(l.s) przy [A] = 1,0 mol/l. Oblicz stałą szybkości i szybkość reakcji przy [B] = 0,2 mol/l. (odpowiedź)
1-14. W reakcji II rzędu A + B 2D początkowe stężenia substancji A i B wynoszą odpowiednio 0,5 i 2,5 mol/l. Ile razy szybkość reakcji przy [A] = 0,1 mol/l jest mniejsza niż szybkość początkowa? (odpowiedź)
1-15. Szybkość reakcji w fazie gazowej opisuje równanie w = k. [A] 2 . [B]. Przy jakim stosunku stężeń A i B początkowa szybkość reakcji będzie maksymalna przy ustalonym ciśnieniu całkowitym? (odpowiedź)
2. Kinetyka reakcji prostych
W tej części, w oparciu o prawo działania mas, ułożymy i rozwiążemy równania kinetyczne dla nieodwracalnych reakcji całego rzędu.
Reakcje 0-go rzędu. Szybkość tych reakcji nie zależy od stężenia:
![]() ,
,
gdzie [A] jest stężeniem substancji wyjściowej. Porządek zerowy występuje w reakcjach heterogenicznych i fotochemicznych.
Reakcje pierwszego rzędu. W reakcjach typu A – B szybkość jest wprost proporcjonalna do stężenia:
![]() .
.
Przy rozwiązywaniu równań kinetycznych często stosuje się następujący zapis: stężenie początkowe [A] 0 = A, aktualne stężenie [A] = A - X(T), Gdzie X(T) to stężenie przereagowanej substancji A. W zapisie tym równanie kinetyczne reakcji I rzędu i jej rozwiązania ma postać:
Rozwiązanie równania kinetycznego zapisuje się również w innej formie, wygodnej do analizy rzędu reakcji:
![]() .
.
Czas, w którym połowa substancji A ulega rozpadowi, nazywany jest okresem półtrwania t 1/2. Jest to określone równaniem X(t 1/2) = A/2 i równe
Reakcje drugiego rzędu. W reakcjach typu A + B D + ... szybkość jest wprost proporcjonalna do iloczynu stężeń:
![]() .
.
Początkowe stężenia substancji: [A] 0 = A, [B] 0 = B; aktualne stężenia: [A] = A- X(T), [B] = B - X(T).
Rozwiązując to równanie, rozróżnia się dwa przypadki.
1) identyczne stężenia początkowe substancji A i B: A = B. Równanie kinetyczne ma postać:
![]() .
.
Rozwiązanie tego równania zapisuje się w różnych postaciach:
Okresy półtrwania substancji A i B są takie same i równe:
2) Początkowe stężenia substancji A i B są różne: A
B. Równanie kinetyczne ma postać: ![]() .
.
Rozwiązanie tego równania można zapisać w następujący sposób:
Okresy półtrwania substancji A i B są różne: ![]() .
.
Reakcje n-tego rzędu N A D + ... Równanie kinetyczne ma postać:
![]() .
.
Rozwiązanie równania kinetycznego:
 . (2.1)
. (2.1)
Okres półtrwania substancji A jest odwrotnie proporcjonalny do ( N-1) stopień początkowego stężenia:
![]() . (2.2)
. (2.2)
Przykład 2-1. Okres półtrwania radioaktywnego izotopu 14 C wynosi 5730 lat. Podczas badań archeologicznych odnaleziono drzewo, którego zawartość 14 C wynosiła 72% normy. Ile lat ma drzewo?
Rozwiązanie. Rozpad promieniotwórczy jest reakcją pierwszego rzędu. Stała szybkości wynosi:
Czas życia drzewa można wyznaczyć rozwiązując równanie kinetyczne, biorąc pod uwagę fakt, że [A] = 0,72. [A] 0:
Przykład 2-2. Ustalono, że reakcja drugiego rzędu (jeden odczynnik) przebiega w 75% w ciągu 92 minut przy początkowym stężeniu odczynnika 0,24 M. Po jakim czasie w tych samych warunkach stężenie odczynnika osiągnie wartość 0,16 M?
Rozwiązanie. Zapiszmy dwukrotnie rozwiązanie równania kinetycznego reakcji II rzędu z jednym odczynnikiem:
![]() ,
,
gdzie, pod warunkiem, A= 0,24 M, T 1 = 92 minuty, X 1 = 0,75. 0,24 = 0,18 M, X 2 = 0,24 - 0,16 = 0,08 M. Podzielmy jedno równanie przez drugie:
Przykład 2-3. Dla elementarnej reakcji N A B oznaczamy okres półtrwania A przez t 1/2, a czas zaniku A przez 75% przez t 3/4. Udowodnić, że stosunek t 3/4 / t 1/2 nie zależy od początkowego stężenia, ale jest określony jedynie przez rząd reakcji N.Rozwiązanie. Zapiszmy rozwiązanie równania kinetycznego reakcji dwukrotnie N-ta kolejność z jednym odczynnikiem:
i podziel jedno wyrażenie przez drugie. Stałe k I A oba wyrażenia zostaną anulowane i otrzymamy:
![]() .
.
Wynik ten można uogólnić, udowadniając, że stosunek czasów, dla których stopień konwersji wynosi aib, zależy tylko od rzędu reakcji:
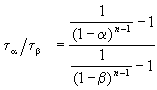 .
.
ZADANIA
2-1. Korzystając z rozwiązania równania kinetycznego wykaż, że dla reakcji I rzędu czas t X, podczas którego osiąga się stopień przemiany substancji wyjściowej X, nie zależy od stężenia początkowego. (odpowiedź)2-2. Reakcja pierwszego rzędu przebiega z szybkością 30% w ciągu 7 minut. Po jakim czasie reakcja zakończy się w 99%? (odpowiedź)
2-3. Okres półtrwania radioaktywnego izotopu 137 Cs, który dostał się do atmosfery w wyniku awarii w Czarnobylu, wynosi 29,7 lat. Po jakim czasie ilość tego izotopu będzie mniejsza niż 1% wartości początkowej? (odpowiedź)
2-4. Okres półtrwania radioaktywnego izotopu 90 Sr, który przedostaje się do atmosfery podczas prób jądrowych, wynosi 28,1 lat. Załóżmy, że organizm noworodka wchłonął 1,00 mg tego izotopu. Ile strontu pozostanie w organizmie po a) 18 latach, b) 70 latach, jeśli założymy, że nie jest on wydalany z organizmu? (odpowiedź)
2-5. Stała szybkości reakcji pierwszego rzędu SO 2 Cl 2 = SO 2 + Cl 2 wynosi 2,2. 10 -5 s -1 w 320 o C. Jaki procent SO 2 Cl 2 ulegnie rozkładowi przy przetrzymywaniu w tej temperaturze przez 2 godziny? (odpowiedź)
2-6. Stała szybkości reakcji pierwszego rzędu
2N 2 O 5 (g) 4NO 2 (g) + O 2 (g)
w 25 o C wynosi 3,38. 10 -5 s -1 . Jaki jest okres półtrwania N 2 O 5? Jakie będzie ciśnienie w układzie po a) 10 s, b) 10 min, jeśli ciśnienie początkowe wynosiło 500 mm Hg? Sztuka. (odpowiedź)
2-7. Reakcję pierwszego rzędu prowadzi się z różnymi ilościami materiału wyjściowego. Czy styczne do początkowych odcinków krzywych kinetycznych przetną się w jednym punkcie na osi x? Uzasadnij swoją odpowiedź. (odpowiedź)
2-8. Reakcja pierwszego rzędu A 2B zachodzi w fazie gazowej. Początkowe ciśnienie wynosi P 0 (brak B). Znajdź zależność całkowitego ciśnienia od czasu. Po jakim czasie ciśnienie wzrośnie 1,5 razy w porównaniu do pierwotnego? Jaki jest postęp reakcji do tego czasu? (odpowiedź)
2-9. Reakcja drugiego rzędu 2A B zachodzi w fazie gazowej. Początkowe ciśnienie wynosi P 0 (brak B). Znajdź zależność całkowitego ciśnienia od czasu. Po jakim czasie ciśnienie spadnie 1,5 razy w porównaniu do pierwotnego? Jaki jest postęp reakcji do tego czasu? (odpowiedź)
2-10. Substancję A zmieszano z substancjami B i C w równych stężeniach 1 mol/l. Po 1000 s pozostaje 50% substancji A. Ile substancji A pozostanie po 2000 s, jeśli reakcja ma: a) zero, b) pierwszy, c) drugi, c) trzeci ogólny porządek? (odpowiedź)
2-11. Która z reakcji – pierwszej, drugiej czy trzeciej – zakończy się szybciej, jeśli początkowe stężenia substancji wynoszą 1 mol/l, a wszystkie stałe szybkości wyrażone w mol/l i s są równe 1? (odpowiedź)
2-12. Reakcja
CH 3 CH 2 NO 2 + OH - H 2 O + CH 3 CHNO 2 -
ma drugiego rzędu i stałą szybkości k= 39,1 l/(min mol.) w temperaturze 0 o C. Przygotowano roztwór zawierający 0,004 M nitroetan i 0,005 M NaOH. Po jakim czasie przereaguje 90% nitroetanu?
2-13. Stała szybkości rekombinacji jonów H + i FG - (fenyloglioksynianu) w cząsteczkę UFG w temperaturze 298 K jest równa k= 10 11,59 l/(mol. s). Oblicz czas potrzebny do zakończenia reakcji w 99,999%, jeśli początkowe stężenia obu jonów wynoszą 0,001 mol/l. (odpowiedź)
2-14. Szybkość utleniania 1-butanolu przez kwas podchlorawy nie zależy od stężenia alkoholu i jest proporcjonalna do 2. Po jakim czasie reakcja utleniania w temperaturze 298 K zakończy się w 90%, jeśli roztwór początkowy zawierał 0,1 mol/l HClO i 1 mol/l alkoholu? Stała szybkości reakcji wynosi k= 24 l/(mol-min). (odpowiedź)
2-15. W określonej temperaturze 0,01 M roztwór octanu etylu zmydla się 0,002 M roztworem NaOH o 10% w ciągu 23 minut. Po ilu minutach zmydli się w tym samym stopniu za pomocą 0,005 M roztworu KOH? Weź pod uwagę, że ta reakcja jest drugiego rzędu, a zasady są całkowicie zdysocjowane.(odpowiedź)
2-16. Reakcję drugiego rzędu A + B P prowadzi się w roztworze o początkowych stężeniach [A] 0 = 0,050 mol/L i [B] 0 = 0,080 mol/L. Po 1 godzinie stężenie substancji A spadło do 0,020 mol/l. Oblicz stałą szybkości i okresy półtrwania obu substancji.
Aby doświadczalnie określić szybkość reakcji chemicznej, konieczne jest posiadanie danych na temat zmian stężenia substancji początkowych lub końcowych w czasie. Metody, za pomocą których można to zrobić, dzielą się na chemiczny I fizykochemiczne.
Metody chemiczne polegają na bezpośrednim określeniu ilości substancji lub jej stężenia w naczyniu reakcyjnym.
Najczęściej do tych celów wykorzystuje się takie rodzaje analiz ilościowych jak miareczkowanie i grawimetria. Jeśli reakcja przebiega powoli, w celu monitorowania zużycia odczynników pobiera się próbki ze środowiska reakcji w określonych odstępach czasu. Następnie określa się zawartość pożądanej substancji. Na przykład miareczkowanie zasadą określa ilość kwasu w układzie w miarę postępu reakcji
R 1 – COOH + R 2 – OH → R 1 – COO – R 2 + H 2 O
Jeśli reakcja przebiega z dużą szybkością, to pobranie próbki zostaje zatrzymane przez nagłe ochłodzenie, szybkie usunięcie katalizatora, rozcieńczenie lub przejście jednego z odczynników do stanu niereaktywnego.
Chemiczne metody analizy charakteryzują się prostotą, dostępnością i dobrą dokładnością.
W nowoczesnej kinetyce eksperymentalnej najczęściej używają fizykochemiczne metody analizy . Pozwalają kontrolować zmiany stężenia substancji bezpośrednio w trakcie reakcji, bez konieczności jej przerywania i pobierania próbki. Metody te opierają się na pomiarze dowolnej właściwości fizycznej układu, która zmienia się w czasie i zależy od ilościowej zawartości określonego związku w nim; na przykład: ciśnienie (jeśli w reakcji biorą udział gazy), przewodność elektryczna, współczynnik załamania światła, widmo absorpcyjne odczynnika lub produktu reakcji w zakresie ultrafioletu, światła widzialnego lub podczerwieni. Szeroko stosowane są widma elektronowego rezonansu paramagnetycznego (EPR) i jądrowego rezonansu magnetycznego (NMR).
Zastosowanie metod spektralnych opiera się na fakcie, że absorpcja promieniowania elektromagnetycznego jest proporcjonalna do ilości substancji lub jej stężenia w układzie.
Zwykle reakcje bada się eksperymentalnie w układzie zamkniętym (tj. przy stałej objętości), a wyniki przedstawia się graficznie w postaci tzw. krzywa kinetyczna, wyrażający zależność stężenia odczynnika lub produktu reakcji od czasu. Analityczna postać tej zależności nazywa się równanie krzywej kinetycznej. W przeciwieństwie do podstawowego równania kinetycznego, równania krzywych zużycia reagujących substancji (lub akumulacji produktów reakcji) zawierają jako parametry początkowe stężenia składników (C 0) w chwili t = 0.
Z tych równań wyprowadza się wzory umożliwiające obliczenie stałej szybkości reakcji i okres półtrwania(t½) – okres czasu, w którym zużywa się połowę przyjętej substancji wyjściowej, tj. jego stężenie zmniejszy się 2 razy i osiągnie wartość C O /2.
W reakcjach zerowego rzędu stężenie substancji wyjściowej maleje liniowo w czasie (Rys. 37)
Ryż. 37. Zmiana stężenia substancji wyjściowej w czasie w reakcji zerowego rzędu
Matematycznie tę zależność liniową można zapisać w następujący sposób:
Gdziek– stała szybkości, C 0 – początkowe stężenie molowe odczynnika, C – stężenie w czasieT.
Można z niego wyprowadzić wzór na obliczenie stałej szybkości reakcji chemicznej zerowego rzędu.
k = (C 0 – C).
Stałą szybkości rzędu zerowego mierzy się w mol/l ∙ s (mol l -1 · Z -1 ).
Czas połowy konwersji dla reakcji zerowego rzędu jest proporcjonalny do stężenia materiału wyjściowego
Dla reakcji pierwszego rzędu krzywa kinetyczna we współrzędnych C, t ma charakter wykładniczy i wygląda następująco (Rys. 38) Matematycznie krzywa ta jest opisana równaniem
C = C 0 e - kt

Ryż. 38. Zmiana stężenia substancji wyjściowej w czasie w reakcji pierwszego rzędu
W praktyce dla reakcji pierwszego rzędu krzywą kinetyczną najczęściej wykreśla się we współrzędnych ℓnC,t. W tym przypadku obserwuje się liniową zależność ℓnС od czasu (ryc. 39)
ℓnС = ℓnС 0 –kt

Ryż. 39. Zależność logarytmu stężenia odczynnika od czasu wystąpienia reakcji pierwszego rzędu
W związku z tym wartość stałej szybkości i czasu połowy konwersji można obliczyć za pomocą poniższych wzorów
k = ℓn lub k= 2,303ℓg
(przy przejściu z logarytmu dziesiętnego na logarytm naturalny).
Stała szybkości reakcji pierwszego rzędu ma wymiar T –1 , tj. 1/s i nie zależy od jednostek stężenia. Pokazuje proporcję cząsteczek reagujących w jednostce czasu do całkowitej liczby cząsteczek odczynnika w systemie. Zatem w reakcjach pierwszego rzędu równe części pobranej ilości substancji wyjściowej są zużywane w równych okresach czasu.
Drugą charakterystyczną cechą reakcji pierwszego rzędu jest to, że t ½ dla nich nie zależy od początkowego stężenia odczynnika, ale zależy jedynie od stałej szybkości.
Postać równania na zależność stężenia od czasu dla reakcji drugiego rzędu rozważymy tylko w najprostszym przypadku, gdy w akcie elementarnym biorą udział 2 identyczne cząsteczki lub cząsteczki różnych substancji, ale ich początkowe stężenia (C 0) wynoszą równy. W tym przypadku obserwuje się liniową zależność we współrzędnych 1/C,t (rys. 40). Równanie matematyczne tej zależności zostanie zapisane w następujący sposób:

Ryż. 40. Zależność odwrotnego stężenia odczynnika od czasu dla reakcji drugiego rzędu
Stały prędkość oblicza się ze wzoru
i jest mierzony w KM -1 ∙mol -1 , tj. jego wartość liczbowa zależy od jednostek, w jakich mierzy się stężenie substancji.
Okres półtrwania reakcji drugiego rzędu jest odwrotnie proporcjonalny do początkowego stężenia odczynnika
Wynika to z faktu, że szybkość reakcji drugiego rzędu silnie zależy od liczby zderzeń cząsteczek reagujących substancji w jednostce czasu, co z kolei jest proporcjonalne do liczby cząsteczek w jednostce objętości, tj. stężenie substancji. Zatem im większe stężenie substancji w układzie, tym częściej cząsteczki zderzają się ze sobą i krócej połowa z nich będzie miała czas na reakcję.
Reakcje trzeciego rzędu, jak wspomniano wcześniej, są niezwykle rzadkie i nie mają praktycznego znaczenia. Dlatego w związku z tym nie będziemy ich rozważać.
stała szybkości reakcji- to szybkość reakcji chemicznej w warunkach, gdy iloczyn stężeń reagujących substancji wynosi 1 mol/l. Chemia ogólna: podręcznik / A. V. Zholnin Stała szybkości reakcji - współczynnik proporcjonalności w kinetyce różniczkowej... ... Terminy chemiczne
stała szybkości reakcji- - [A.S. Goldberg. Angielsko-rosyjski słownik energii. 2006] Tematy: energia ogólnie EN stała reakcji ...
stała szybkości reakcji- reakcijos greičio konstanta statusas T sritis chemija apibrėžtis Reakcijos, kurios reaguojančiųjų medžiagų koncentracijos lygios vienetui, greitis. atitikmenys: pol. stała szybkości; stała reakcji rus. stała szybkości reakcji; konkretny... ... Chemijos terminų aiškinamasis žodynas
stała szybkości reakcji- reakcijos spartos konstanta statusas T sritis Standartizacija ir metrologija apibrėžtis Reakcijos, kurios reaguojančių medžiagų koncentracijos yra lygios vienetui, sparta. atitikmenys: pol. stała szybkości reakcji vok. Reaktionskonstante, f rus.… … Penkiakalbis aiškinamasis metrologijos terminų žodynas
Główną cechą kinetyczną jest reakcja chemiczna; współczynnik proporcjonalności w równaniu kinetycznym zależności szybkości reakcji od stężeń reagentów i ich współczynników stechiometrycznych. Dla jednocząsteczkowych... ... Wielki słownik encyklopedyczny
stała szybkości reakcji katalitycznej- - [A.S. Goldberg. Angielsko-rosyjski słownik energii. 2006] Ogólne tematy energetyczne EN współczynnik katalityczny ... Przewodnik tłumacza technicznego
Reakcja chemiczna, jej główne cechy kinetyczne; współczynnik proporcjonalności w równaniu kinetycznym zależności szybkości reakcji od stężeń reagentów i ich współczynników stechiometrycznych. Dla jednocząsteczkowych... ... słownik encyklopedyczny
stała szybkości reakcji chemicznej- zmiana ilości (stężenia) substancji, która reaguje lub powstaje w trakcie procesu w jednostce czasu, przy danej temperaturze i stężeniach wszystkich składników równych jedności: d[A]/dt =… … Encyklopedyczny słownik metalurgii
Chem. reakcja, jej główna kinetyka. Charakterystyka; współczynnik proporcjonalność w kinetyce. równanie łączące szybkość reakcji ze stężeniami reagentów i ich stechiometrią. współczynniki. W przypadku reakcji jednocząsteczkowych K. s. ma wymiar z... Naturalna nauka. słownik encyklopedyczny
Względne stałe szybkości reakcji CH 3 I + Cl - w różnych rozpuszczalnikach w temperaturze 25 °C (wg Parkera)- Rozpuszczalnik Względna stała szybkości CH3OH 1 HCONH2 12,5 HCONHCH3 … Podręcznik chemiczny
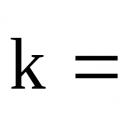 Stała szybkości reakcji Wytyczne dotyczące prac laboratoryjnych
Stała szybkości reakcji Wytyczne dotyczące prac laboratoryjnych Czy istnieje życie po śmierci?
Czy istnieje życie po śmierci? Naukowcy: Ziemia jest „kosmicznym więzieniem” dla ludzkości. Naukowcy: Ziemia jest kosmicznym więzieniem we wszechświecie
Naukowcy: Ziemia jest „kosmicznym więzieniem” dla ludzkości. Naukowcy: Ziemia jest kosmicznym więzieniem we wszechświecie