They do not have a crystalline structure. Crystal structure
The above allows us to give the following definition to the concept of “crystal structure”. A crystalline structure is a structure characterized by an ordered arrangement of particles at strictly defined points in space that form a crystal lattice. This ordering allows us to experimentally and theoretically fully study the structure of the solid state and phenomena associated with the nature of interaction forces in crystalline bodies.
Each crystal has a characteristic anisotropy and a pronounced temperature of transition to the liquid state. Crystals are characterized by external symmetry in the arrangement of particles, which is expressed by the presence of three symmetry elements: center, axis and plane of symmetry. Center of symmetry – a point dividing in half all straight lines connecting the outer surfaces of the crystal, drawn through it in any direction. Plane of symmetry divides the crystal into two parts, related to each other, like an object to its mirror image. Axis of symmetry- this is a line, when rotated around which by a certain angle, the new position completely coincides with the previous one. The more symmetry elements, the higher the external symmetry of the crystal. A perfectly symmetrical figure is a ball.
Currently, the entire variety of crystalline forms by combination of symmetry elements (systemony) is reduced to seven types: regular (cubic), trigonal, hexagonal, tetragonal, rhombic, monoclinic and triclinic. Table 3.2. The classification of crystals by system is given.
Table 3.2. Classification of crystals by system
Crystals of the lower system are characterized by less symmetry; crystals of a higher category of system have a more perfect shape of the crystal lattice and, therefore, are more stable under certain conditions of existence.
Many substances in the crystalline state are characterized by polymorphism, i.e. the ability of a substance to exist in the form of several crystalline structures with different properties. Polymorphism of simple substances is called allotropy. Polymorphic modifications of carbon (diamond, graphite), quartz (α-quartz, β-quartz), iron, tungsten, etc. are known.
If two different substances have the same crystal structure, similar chemical formula, and are not very different in the size of their constituent particles, then they can form mixed crystals. Such substances are called isomorphic; their ability to form mixed crystals is called isomorphism. Example: kaolinite Al 2 O 3 crystals are similar in composition and structure, but different in properties. 2SiO2. 2H 2 O, pyrophyllite Al 2 O 3. 4SiO2. 2H 2 O and montmorillonite Al 2 O 3. 4SiO2. 3H2O.
Real crystals. In our practical activities, we deal with real crystals, which differ from ideal ones by disturbances (defects) of the crystal lattice, formed as a result of changes in the equilibrium conditions of crystal growth, the capture of impurities during crystallization, and also under the influence of various kinds of external influences.
Amorphous structure
Amorphous structure is one of the physical states of solids. Amorphous substances are characterized by two features. Firstly, the properties of such substances under normal conditions do not depend on the chosen direction, i.e. They - isotropic. Secondly, with increasing temperature, the amorphous substance softens and gradually transforms into a liquid state. The exact melting point is not available.
Common to the crystalline and amorphous states of substances is the absence of translational movement of particles and the preservation of only their oscillatory motion around the equilibrium position. The difference between them is the presence of a geometrically regular lattice in crystals and the absence of long-range order in the arrangement of atoms in amorphous substances.
The amorphous state of a substance, compared to the crystalline one, is always less stable and has an excess supply of internal energy. In this regard, under certain conditions, a transition from the amorphous to the crystalline state occurs spontaneously.
Solids in the amorphous state can be obtained in two ways. The first way is rapid cooling of melts of crystalline substances, mainly of ionic and covalent structure. Typical representatives of such amorphous bodies are silicate glasses, bitumen, resins, etc.
The second way is the dispersion of crystal structures. As a result of the dispersion of crystalline bodies, amorphized dispersions are formed in the form of colloids and solutions. By collapsing or condensing, dispersions change their state of aggregation. Supersaturated solutions, for example, can gel and form a polymer or crystallize.
Amorphous substances are divided into vitroids (glasses), dispersed systems and polymers.
Vithroids- These are solids in an amorphous state with a glassy structure. As already noted, glasses are formed as a result of rapid cooling, mainly of silicate melts. Rapid cooling prevents the creation of an ordered structure. Especially if the molecules are bulky and the cooling rate is high.
Lecture 10
Structure of solids. Principles for describing crystal structures
Most of the substances around us are in a solid state. Some solids are shiny and can be deformed when cold - they are classified as metals Others are crystals with regular crystal faces and clear cleavage planes, some of them are classified as salts, or ionic crystals, and some to covalent crystals. A number of other solids are soft and retain many of the properties of the molecules of the gas from which they condensed - these are molecular crystals.
The observed crystal structure of solids (the arrangement of atoms in a unit cell) is determined by the position of the minimum energy of the system as a function of the coordinates of the centers of the atoms. In general, this minimization requires quantum chemical calculations for a set of possible atomic positions and is thus a rather complex mathematical procedure. However, in a number of cases (with non-directional unsaturated forces of interatomic interactions - in purely ionic, van der Waals or metal crystals), the description of the structure of crystals can be significantly simplified if we consider atoms as rigid balls with certain (characteristic of a given atom in a given charge state for a given type of interatomic interaction) radii. This approach, despite its obvious approximation, as was shown earlier, for crystals with ionic and van der Waals bonds can be justified by a sharp increase in the energy of mutual repulsion when atoms approach each other to the state of noticeable overlapping of electronic shells and the smallness of this energy at large interatomic distances.
Solids, unlike liquid and gaseous ones, are characterized by resistance to shear deformation, which allows the substance to maintain its shape under the influence of external forces. This feature is closely related to the long-range nature of interatomic interaction, leading to an ordered arrangement of particles (atoms, molecules or ions) that make up a solid. Maximum degree of order - long range order i.e., strictly periodic repetition of the correct arrangement of particles at any point of a solid body is realized in crystals, whereas amorphous solids are characterized only closer1st order- regular arrangement of particles at distances not exceeding several interatomic ones. As a consequence, the transition of amorphous solids to the liquid state, unlike crystals, occurs continuously, and in this sense, amorphous bodies (for example, glasses) are sometimes considered as supercooled liquids.
Let's consider three classes of substances: molecular crystals, covalent crystals and metals.
Figure 1 shows which of these classes the crystals of the elements of the periodic table belong to. There are only 15 elements that clearly form molecular crystals (top right of the table), and about 70 metal elements (left of the table). Between metals and molecular crystals are elements that include covalent crystals, as well as some solids that are difficult to classify. Some elements (such as arsenic and antimony) have both molecular and metallic forms. Phosphorus also produces both covalent and molecular crystals. These boundary elements are especially important due to their intermediate nature, and we will pay special attention to them.
Compounds of two different nonmetals always form molecular or covalent crystals. The combination of a metal and a nonmetal usually forms an ionic or covalent crystal. Two metals can form one or more metal compounds or (more often) a series of metal solutions, where one element is dissolved in another.
The regularities of the structure of non-metallic crystals are described by rule (8-N) Hume-Rothery, according to which the coordination number of an atom (the number of bonds by which an atom is connected to nearby atoms) CN = 8 - N, Where N‑ group number in the short-period version of the periodic table.
Since the rule is based on ideas about the stability of an electronic octet and an electron pair performing a single covalent bond, the rule is valid only for elements of the main subgroups starting from group IV.
For example, in crystals of elements of the 6th group (S, Se) CN = 8 - 6 = 2; thus, the structure will contain either ring molecules (S8 in orthorhombic and monoclinic sulfur) or long polymer chains (S¥ in plastic sulfur and selenium). Atoms in rings and chains are interconnected by covalent bonds, and van der Waals forces act between the chains and rings.
For crystals of group VII elements, CN = 8-7 = 1, which leads to a crystal consisting of diatomic molecules, for example I2. The atoms in a molecule are connected by covalent bonds, and the molecules are combined into a crystal by van der Waals forces.
However, not all elementary crystals have a molecular type of bond. It can be seen that only in the case of elements of the IV main subgroup (more precisely, C, Si, Ge) can crystals with exclusively covalent bonds be formed, since the coordination number is 4, which is derived from rule 8 - N, enough to link all the atoms of a crystal with a three-dimensional network of covalent bonds. The main feature of such elementary crystals is their tendency to polymorphism and, as a consequence, the variety of properties they exhibit (Figure 2). Stable modifications of these elements are covalent crystals with high values of mechanical characteristics (Young's modulus, shear modulus, strength, hardness), as well as high melting and boiling points. A typical example: silicon, in the crystal lattice of which (also called the diamond lattice) each atom in a state of sp3 hybridization is surrounded by a tetrahedron of neighboring silicon atoms. Such a rigid three-dimensional network of tetrahedral bonds provides high stability to the crystal lattice. Crystalline silicon has a high melting point (1420 °C) and boiling point (3300 °C), exceptional strength and chemical resistance (insoluble in water and acid solutions).
We have already said that most simple and complex substances under normal conditions are solids. One of the most important tasks of solid state chemistry is to establish the relationship between the structure of solids and their properties.
Let me remind you that crystal structure is the specific arrangement of atoms in a crystal. This location is averaged over time and space and corresponds to the average statistical maxima of the electron or nuclear density of the crystal.
An idealized mathematical form of the arrangement of atoms in a crystal, described by a set atomic positions within crystal lattice and one of 230 space symmetry groups, corresponds ideal structure. Distinguish completely ordered structures, in which each atomic position is completely occupied by atoms of the same type, and disordered structures, where there are atomic positions that are not completely occupied by single-sort atoms. Different aspects of the crystal structure are considered within different structure models. Local features microstructures crystal meet real structure. The crystal structure is determined experimentally by methods structural analysis.
The crystal structure (internal structure) determines the multifaceted shape of the crystal (external structure).
A crystal is a solid body characterized by the presence of both short- and long-range order. This is the equilibrium form of the solid state of a substance.
All crystals, without exception, are characterized by a lattice structure. To imagine such a lattice, mentally fill the space with many equal parallelepipeds, parallel oriented and touching along entire faces. The simplest example of such a building is a masonry of bricks closely adjacent to each other. If we select the corresponding points inside each parallelepiped (for example, their centers of gravity or vertices), we obtain a model spatial lattice. In specific crystal structures, the sites of spatial lattice nodes can be filled by individual atoms or ions, or groups of atoms - molecules. The straight lines along which particles are located in the lattice are called in rows, and the planes seated with particles are called flat meshes. Flat grids, rows, vertices correspond to the faces and edges of the crystal.
A crystal lattice is a three-dimensional arrangement of material particles (atoms, ions, molecules) that make up a crystal.
Conventionally, the equivalence of coordinate directions can be shown in the form of unit vectors - scales a, b, c - along the corresponding coordinate axes X, Y, Z.
Three possibilities for the relationship of unit vectors - a = b = c, a = b ≠ c, a ¹ to ¹ c - allow us to divide crystallographic coordinate systems into three groups - three categories of crystals:
· crystals of the highest category(a = b = c) are characterized by complete equivalence of the coordinate axes, which is due to the presence of several higher-order axes in the symmetry groups of such crystals;
· medium grade crystals(a = b ≠ c) are characterized by partial equivalence of coordinate axes, associated with the presence in their groups of only one axis of higher order;
· low grade crystals(a ≠ b ≠ c) are characterized by complete non-equivalence of coordinate directions, which is explained by the absence of higher order axes in them.
Having considered the angular relationships in each of the listed categories, we can derive all crystallographic coordinate systems (systems).
Symmetry classes with a single coordinate frame are combined into a family called syngony or system.
There are a total of 32 different crystallographic point groups, which include different combinations of closed symmetry operations. These point groups are classified according to their affiliation with crystallographic systems.
Crystallographic | Relationships between edges of a unit cell | Point groups |
Triclinic | a ≠ b ≠ c a ≠ b ≠ g ≠ 90° | |
Monoclinic | a ≠ b ≠ c a = b = 90° ≠ g | 2, (only in one direction along the Z axis). m |
Orthogonal (orthorhombic) | a ≠ b ≠ c a = b = g = 90° | |
Tetragonal |
a = b = g = 90° | 4, , 4/m, 422, 4mm, 2m, 4/mmm (the 4th order axis runs only along the Z axis) |
Trigonal and hexagonal |
6, , 6/m, 622, 6mm, m2, 6/mmm (the 3rd (6th) order axis runs only along the Z axis) |
|
Cubic |
a = b = g = 90° | 23, m3, 432, 4m, m3m (4 3rd order axes pass along the volumetric diagonals of the unit cell) |
A system of equivalent positions is a set of points that are translated into each other by the symmetry elements of a given point group.
The number of points included in a given SEP is called the system multiplicity or the position multiplicity. Or - multiplicity is the number of points obtained by multiplying their symmetry elements.
The position is called private, if the point is located on any element of symmetry: on an axis, on a plane of symmetry, at a special point of inversion, at the center of symmetry.
The position is called general, if the point is not on the symmetry element.
Atoms in a crystal can be connected not only by closed symmetry elements, but also by open symmetry elements.
The set of three non-coplanar vectors is called the translation group or crystal lattice.
Vectors a,b, c are called transfer vectors or translations, and their modules are called lattice identity periods.
A parallelepiped built on vectors a,b, s, is called a parallelepiped of lattice repetition.
The constituent elements of a lattice are its nodes, nodal rows and nodal meshes.
A spatial lattice is a geometric image that reflects the three-dimensional periodicity of the distribution of atoms in the structure of a crystal.
The lattice is defined by one type of atom.
To define a lattice, you need to select a parallelepiped that would most fully reflect all the features of a given lattice, being its minimum link, i.e. you need to select an elementary cell.
The unit cell is a parallelepiped of repetition, built on the shortest translations along crystallographic coordinate systems.
Three possible vector relationships – a = b = c, a = b ¹ c, a ¹ b ¹ c– make it possible to divide crystallographic coordinate systems, and therefore 32 symmetry classes, into three categories of crystals:
1. crystals of the lowest category (a ¹ b ¹ c) are characterized by complete non-equivalence of coordinate directions, which is explained by the absence of higher order axes in them. From the condition of non-equivalence of coordinate directions it follows that the lowest category includes only classes that do not have axes of higher order..gif" width="13" height="20 src=">) or the complete absence of symmetry elements (1).
2. medium category crystals (a = b ¹ c) are characterized by partial equivalence of coordinate axes, associated with the presence in their symmetry groups of only one higher order axis. From the condition of equivalence of two horizontal directions (a = b) it follows that the symmetry of crystals of the middle category is described by groups with a single axis of higher order: . The vertical coordinate axis is combined with this axis z, and the other two – x And y– selected in a plane perpendicular to the main axis. Therefore, the angles between the main axis and the axes x And y straight, i.e. a = b = 90°. Angle g between axes x And y is determined by the order of the main axis and is equal to 90° in the case of the presence of an axis of the 4th order and 120° in the case of axes of the 3rd and 6th orders. Therefore, in the middle category there are two coordinate systems, which correspond to two systems.
3. crystals of the highest category (a = b = c) are characterized by complete equivalence of coordinate axes, which is due to the presence of several higher order axes in their symmetry groups.
So, on these three translations it is possible to construct an elementary parallelepiped - an elementary cell. Options a, b, c, a, b, g- unit cell parameters.
Let me remind you rules for choosing a unit cell (rules for choosing crystallographic coordinate axes).
1. The selected cell must have lattice symmetry.
2. Crystallographic axes are directed along the nodal rows.
3. Crystallographic coordinate axes are combined with special directions, i.e., with symmetry axes of the 2nd order and higher (if any).
4. All other things being equal, the unit cell must have a minimum volume.
If you select any point in three-dimensional space (not necessarily a material one) and consider it one of the lattice nodes, then in its remaining nodes there will be all points of this space that are identical (physically and geometrically) to the original one.
In this sense lattice‑it is an expression of the crystalline state of a substance, for any crystalline substance, even devoid of any other elements of symmetry, always possesses this basic element of symmetry - a lattice, or lattice structure.
Like any parallelepipedal system, a three-dimensional lattice has a number of its own symmetry features. It is always centrosymmetric, and the centers of inversion are located both at lattice nodes - at the vertices of parallelepipeds, and at the midpoints of the distances between them. Axes of higher orders are inevitably accompanied by planes of symmetry intersecting along them. The symmetry axes themselves are limited only by crystallographic orders, i.e. n= 1, 2, 3, 4, 6. The last condition uniquely selects from the infinite number of point groups that describe the symmetry of the final initial figures, only 32 crystallographic point groups.
The point symmetry groups of the lattice as a geometric image correspond to the highest- holohedral-class of each system.
A three-dimensional lattice can be represented by three non-coplanar translation vectors, which means a parallelepiped built on these vectors is repetition parallelepiped- will lattice cell. In order for a parallelepiped to serve as a characteristic cell of any lattice, i.e., to reflect its main symmetry features, it is necessary that its edges (translation vectors) coincide with the special directions of maximum symmetry, i.e., with the directions of the crystallographic coordinate axes. A cell selected in this way is called Bravais cell or unit cell. The type and symmetry of the cell are reflected in its name, which it conveys and the corresponding spatial lattice (Figure 3). Since the shape of the Bravais cell is determined by the coordinate reference, seven lattices of different symmetry (, , mmm, https://pandia.ru/text/80/189/images/image013_92.gif" width="46" height="41 src="> .gif" width="14" height="19 src=">m) can be represented by six types of parallelepipeds (since hexagonal lattices are served by the same coordinate reference, and therefore by Bravais cells of the same shape - parallelepipeds with a 120-degree rhombus at the base).
To characterize the type of lattice, it is necessary and sufficient to indicate two of its characteristics:
1. crystallographic system;
2. type of “centering” of the cell.
Unit cells can be:
1. primitive - only the vertices of the cell are nodes;
2. centered – there are additional nodes that do not lie at the vertices of the cell.
Figure 3 – Types of Bravais gratings
If the crystallographic axes are chosen correctly, then additional nodes are not possible in any place, but only in strictly defined positions. However, the number of possible options is small. Non-primitive lattices are called centered.
Non-primitive (centered) lattices can be of the following type:
I - body-centered (the node is in the center of the volume)
C (A, B) – base-centered (two opposite faces are centered)
F – face-centered (additional nodes are located at the centers of all faces)
R – double body-centered (two additional nodes divide the volume diagonal into three equal parts)
The rules that determine the choice of coordinate systems in groups of different crystallographic systems (systems) also limit the ways of centering their lattices in different ways.
To describe the symmetry of crystal structures, the concept of “space group” is used.
The set of symmetry elements of a crystal structure is called a space group.
To describe a structure means to indicate:
2) Bravais lattice type;
3) type of chemical formula;
4) CN and coordination polyhedra;
5) number of formula units, etc.
6) characterization of the structure by type of chemical bond;
7) characteristics of the structure based on geometric characteristics;
8) structure in terms of PShU-PSK;
9) basic coordinates of atoms;
10) space group and structural type.
The structure of metals, along with the structure of non-metallic elementary crystals, is presented in Figure 4.
The bottom line of each square shows the form that is stable at room temperature, followed by the forms that occur at higher temperatures.
The abbreviation fcc denotes a face-centered cubic structure with the closest packing of atoms, bcc - a body-centered cubic structure, hcp - a hexagonal structure with a close packing of atoms.
The fcc and hcp structures are most clearly described within the framework of the closest spherical packing (PU) model, first proposed in 1926 by W. Goldschmidt. Atoms are represented as rigid balls, and in the plane there is only one option for their dense arrangement (Figure 5 a).
If the second layer is placed so that its balls are located in the recesses of the first layer, then the densest packing is completed, and also in the only possible way (Figure 5 b). As for the balls of the third layer, they can be arranged in two ways:
1) balls of the third layer above the balls of the first, balls of the fourth above the balls of the second, etc., so that the alternating layers correspond to the sequence ABAVABAB (where the letters A and B indicate densely packed layers, shifted relative to each other in the horizontal plane), and the laying method corresponds hexagonal close packing (hcp) (Figure 6, A);
2) the balls of the third layer in relation to the balls of the second layer are located so that they are not above the balls of the first layer.
Then the fourth layer repeats the first, the second repeats the fifth, etc. The alternation of layers corresponds to ABCAB... ABC, and the laying method corresponds to cubic close packing (FCC) (Figure 6, b). In the hcp structure, the packing of balls in the plane of the layer and vertically to it are different, but in the fcc structure, the packing is the same in any of the three main planes of the cube (i.e., it is less anisotropic). Despite their differences, these two types of dense spherical packings exhibit common features:
1) the proportion of space occupied by the balls - the filling factor, in both cases is equal to 74.05%;
2) the coordination number of the atom is 12;
3) in both packages there are two types of voids - tetrahedral, formed by four contacting balls, and octahedral, respectively, formed by six balls; a tetrahedral void can accommodate a ball with a radius rtetra = 0,225 r, and into an octahedral ball with radius rOct=0,414 r, Where r- radius of the balls that make up the densest packing;
4) in the closest packings, per one ball there is one octahedral and two tetrahedral voids.
In the concept of closest packing, polymorphism is considered as an order of alternation of close-packed layers, different from fcc and hcp.
An example is the sequence of layers in a four-layer hexagonal packing...ABSBABBC... (denoted as 4H).
Of the spherical packings with a lower density, the most common is body-centered cubic packing (BCC), for which the fill factor is 68.01%.
This type of packaging can be obtained if balls of the same size are placed on a plane so that their square arrangement is formed, then the balls of the second layer should be placed in the recesses formed by the balls of the first layer (Figure 7), the balls of the third layer will repeat the first, etc. As in the case of the hcp structure, the alternation of layers corresponds to the sequence ABAB...AB, however, each of the layers is not close-packed; Unlike fcc and hcp, where the coordination number of atoms is 12, the structure under consideration has a coordination number of 8.
|
Figure 7 – Body-centered cubic lattice |

It is easy to see that metals are characterized by polymorphism (allotropy) (Figure 2), and a slight change in the electronic structure of atoms is sufficient for a restructuring of the crystal lattice to occur. The heat of mutual transition between the fcc and hcp structures does not exceed 1 kJ/mol, while the heat of fusion ranges from 10 to 40 kJ/mol.
The vast majority of metals have one of three structures (fcc, bcc, hcp), and Mn, Ga, In, Hg are similar, but distorted structures. It is difficult to detect simple patterns between the type of structure and the position of the metal in the periodic table. Nevertheless, it is obvious that an increase in the number of unpaired valence s- And R-electrons in the state used to form a bond from 1 (alkali metals) to 3 (metals of the third main subgroup), increases the CN from 8 (bcc lattice) to 12 (fcc or hcp lattice). In polymorphism, the effect of increasing the number of valence electrons is equivalent to decreasing the temperature or increasing the pressure.
All non-metallic elements except oxygen are diamagnetic. Metals, with the exception of those belonging to groups 1B-IIIB, are paramagnetic. Among metals, iron, cobalt and nickel have exceptionally high magnetism. Based on the type of temperature dependence, the following groups of metals can be distinguished: magnetic properties remain almost unchanged up to 1100 °C (Mo, W, Os); magnetic susceptibility obeys the Curie-Weiss law (K, Mg, Zn, In, Sc); magnetic properties change slightly at the melting temperature (Na, Cd, A1); with an anomalous change in magnetic properties (Ag, Au, Tl, Sn, Pb, Sb, Bi) and, finally, magnetic properties change (Zn, Tl) or do not change (Ti, Sn) at the transition points. The set of chemical elements, ordered in accordance with Mendeleev's periodic law, is divided into subsets, i.e., fairly isolated areas of chemical elements corresponding to typical metals, ferromagnets, superconductors, dielectrics, semiconductors and semimetals.
1.4. Main types of crystal structures
The point arrangement of atoms in spatial lattices is simplified and unsuitable for studying crystal structures, when the distance between nearest atoms or ions is determined. However, the physical properties of crystalline structures depend on the chemical nature of the substances, the sizes of atoms (ions) and the interaction forces between them. Therefore, in what follows we will assume that atoms or ions have the shape of a ball and are characterized by effective radius, meaning by it the radius of their sphere of influence, equal to half the distance between the two nearest neighboring atoms or ions of the same type. In a cubic lattice, the effective atomic radius is a 0 /2.
The effective radius has different eigenvalues in each particular structure and depends on the nature and number of neighboring atoms. The atomic radii of different elements can only be compared when they form crystals with the same coordination number. Coordination number z of a given atom (ion) is the number of surrounding nearest atoms (ions) of the same type in the crystal structure. Mentally connecting the centers of neighboring particles with straight lines to each other, we get
coordination polyhedron; in this case, the atom (ion) for which such a polyhedron is constructed is located in its center.
The coordination number and the ratio of the effective radii of particles are related to each other in a certain way: the smaller the difference in particle sizes, the larger z.
Depending on the crystal structure (lattice type), z can vary from 3 to 12. As will be shown below, in the structure of diamond z = 4, in rock salt z = 6 (each sodium ion is surrounded by six chlorine ions). For metals, a typical coordination number is z = 12, for crystalline semiconductors z = 4 or z = 6. For liquids, the coordination number is determined statistically as the average number of nearest neighbors of any atom.
The coordination number is related to the packing density of the atoms in the crystal structure. Relative packing density–
This is the ratio of the volume occupied by atoms to the total volume of the structure. The higher the coordination number, the higher the relative packing density.

Section 1. Basic principles of physicochemical crystallography
The crystal lattice strives to have a minimum of free energy. This is only possible if each particle interacts with the maximum possible number of other particles. In other words, the coordination number should be maximum m. The desire for dense packing is characteristic of all types of crystal structures.
Let us consider a flat structure consisting of atoms of the same nature that touch each other and fill most of the space. In this case, only one way is possible for the densest packing of atoms adjacent to each other: around the central
the centers of gravity fall on the voids of the first layer. This can be clearly seen in the right image in Fig. 1.10, a (top view), where the projections of the atoms of the second layer are colored pale gray. The atoms of the second layer form a basic triangle (shown as a solid line) with the apex pointing upward.
Rice. 1.10. The sequence of layers when packing balls of the same size in structures of two types: a – ABAB... with hexagonal close packing (hcp); b – ABCAVS... in the densest cubic package (K PU), giving a face-centered cubic (FCC) lattice. For clarity, the third and fourth layers are shown not completely filled

Chapter 1. Elements of crystal physics
The atoms of the third layer can be arranged in two ways. If the centers of gravity of the atoms of the third layer are above the centers of gravity of the atoms of the first layer, then the laying of the first layer will be repeated (Fig. 1.10, a). The resulting structure is hexagonal close packing(GPU). It can be represented as a sequence of layers ABAVABAB ... in the direction of the Z axis.
If the atoms of the third layer C (shown in dark gray on the right in Fig. 1.10, b) are located above other voids of the first layer and form a basic triangle, rotated 180º relative to layer B (shown by a dotted line), and the fourth layer is identical to the first, then the resulting structure represents cubic dense packing(FCC), which corresponds to a face-centered cubic structure (fcc) with a sequence of layers АВСАВСАВСАВС... in the direction of the Z axis.
For the densest packings z = 12. This is clearly seen in the example of the central ball in layer B: its immediate surroundings are six balls in layer A and three balls below and above it in layers B
(Fig. 1.10, a).
In addition to the coordination number z, various structures are also characterized by packing density, introduced as the ratio of the volume V at occupied by atoms to the volume of the entire Bravais cell V cell. Atoms are represented as solid balls of radius r, therefore V at = n (4π/3)r 3, where n is the number of atoms in the cell.
The volume of a cubic cell is V cell = a 0 3, where a 0 is the lattice period. For an HPU cell with a hexagonal base area S = 3a 0 2 2 3
and height c = 2a 0 23 we obtain V cell = 3a 0 3 2 .
The corresponding parameters of crystal structures - primitive cubic (PC), body-centered cubic (BCC), face-centered cubic (FCC), hexagonal close-packed (HCP) - are given in Table. 1.2. The radii of the atoms are written taking into account the fact that they touch along the edges of the cube in the PC structure (2r = a 0), along the spatial diagonals (4r = a 0 3) in the bcc structure and along the diagonals of the faces (4r = a 0 2)
in the fcc structure.
Thus, in structures with the closest packing (fcc and hcp), having z = 12, the volume of the cell is 74% occupied by atoms. As the coordination number decreases to 8 and 6, the packing density decreases to 68 (bcc) and 52% (PC), respectively.
![]()
Table 1.2
Parameters of cubic and hexagonal crystals
Crystal parameters |
|||||
Coordination number z |
|||||
Number of atoms n in a cell |
|||||
Atomic radius r |
a 0 /2 |
a 2 4 |
a 0 /2 |
||
Volume of one atom, V at /n |
|||||
a 0 3 π 6 |
a3 π |
a 3 π 2 24 |
π a 0 3 6 |
||
Packing density |
|||||
π 3 8 = 0.6 |
π 2 6 = 0.74 |
π 2 6 = 0.74 |
|||
V at/ V cell |
|||||
It has already been noted that during the crystallization of a substance, the system strives to provide a minimum of free energy. One of the factors that reduces the potential energy of interaction between particles is their maximum approach and the establishment of mutual communication with the largest possible number of particles, i.e., the desire for denser packing with the highest coordination number.
The tendency towards the implementation of dense packing is characteristic of all types of structures, but it is most pronounced in metallic, ionic and molecular crystals. The bonds in them are non-directional or weakly directed (see Chapter 2), so for atoms, ions
And For molecules, the model of solid incompressible spheres is quite acceptable.
The Bravais translation lattices shown in Fig. 1.3
And in table 1.1, all possible options for constructing crystal structures, primarily for chemical compounds, are not exhausted. The fact is that periodic repetition of the Bravais cell produces a translation lattice consisting only of particles (molecules, atoms, ions) of one type. Therefore, the structure of a complex compound can be built by a combination of Bravais lattices inserted into one another in a certain way. Thus, semiconductor crystals use a directed covalent (non-polar or polar) bond, which is usually realized by combining at least two lattices, individually quite tightly packed, but ultimately providing small coordination numbers of the “total” lattice (up to z = 4).
There are groups of substances characterized by an identical spatial arrangement of atoms and differing from each other only in the parameters (but not the type) of the crystal lattice.

Therefore, their structure can be described using one spatial model ( one structural type) indicating specific values of lattice parameters for each substance. Thus, crystals of various substances belong to a limited number of structural types.
The most common types of structures are:
in metal crystals:
tungsten structure (OC K-lattice); copper structure (fcc lattice), magnesium structure (hcp lattice);
in dielectric crystals:
structure of sodium chloride (double GC lattice); structure of cesium chloride (double PC lattice);
in semiconductor crystals:
diamond structure (double fcc lattice); sphalerite structure (double G CC-lattice); wurtzite structure (double HP Y-lattice).
Let us briefly consider the features and realizability of the structures listed above and the corresponding Bravais lattices.
1.4.1. Metal crystals
Tungsten structure(Fig. 1.1 1, a). A body-centered cubic lattice is not a structure with the closest packing; it has a relative packing density of 0.6 8 and a coordination number z = 8. The planes (11 1) are most densely packed.
Rice. 1.11. Types of cubic lattices: a – body-centered cubic (bcc); b – simple cubic

Section 1. Basic principles of physicochemical crystallography
In addition to tungsten W, all alkali and alkaline earth metals, as well as most refractory metals, have a bcc lattice: chromium Cr, iron Fe, molybdenum Mo, zirconium Zr, tantalum Ta, niobium Nb, etc. The latter has the following explanation. In a bcc cell for the central atom, the nearest neighbors are the atoms at the vertices of the cube (z = 8). They are spaced apart from each other
six central atoms in neighboring cells (second coordination sphere), which practically increases the coordination number to z 14. This gives a total energy gain that compensates for the negative contribution from a small increase in the average distances between atoms compared to the fcc lattice, where the atoms are located at a distance d = a 0 ( 2) 2 = 0.707a 0 . As a result, the strength increases
ity of the crystals, manifested in their high melting point, reaching 3,422 ºC for tungsten. For comparison: a simple cubic structure (Fig. 1.11, b) with z = 8 has loose packing and is found only in polonium Po.
The structure of copper (fcc lattice), shown in Fig. 1.12, a, refers to close-packed structures, has a relative packing density of 0.74 and a coordination number z = 12. In addition to copper Cu, it is characteristic of many metals, such as gold Au, silver Ag, platinum Pt, nickel Ni, aluminum Al, lead Pb, palladium Pd, thorium Th, etc.
Rice. 1.12. Structures of close-packed crystal lattices: a – face-centered cubic (copper structure); b – hexagonal close-packed (magnesium structure)

Chapter 1. Elements of crystal physics
The listed metals are relatively soft and ductile. The fact is that in copper-type structures, the tetrahedral and octahedral voids in the fcc lattice are not filled with other particles. This allows, due to the non-directionality of bonds between atoms, their displacement along the so-called sliding planes. In the fcc lattice, these are the planes of the largest packing (111), one of which is shown shaded in Fig. 1.12, a.
Structure of magnesium(HCP lattice) shown in Fig. 1.12, b, is typical not only for magnesium Mg, but also for cadmium Cd, zinc Zn, titanium Ti, thallium Tl, beryllium Be, etc., as well as for most rare earth elements. Unlike the PC lattice, the GPU lattice in Fig. 1.12, b has layer B (shaded), located in the middle between the base layers A at a fixed distance
with 2 = a 0 2 3 (with an observed deviation of up to 10% for some
other metals). The atoms in the B layers are located above the centers of the triangles in the basal plane (0001) with the closest packing.
1.4.2. Dielectric crystals
Structure of sodium chloride(Fig. 1.13, a) can be described
san as two face-centered cubic lattices (structural type of copper), shifted by half a lattice period (a 0 /2) along any of the edges<100>.
Large chlorine anions Cl− occupy the nodes of the fcc cell and form a cubic close packing, in which the sodium cations Na+, having a smaller size, fill only the octahedral voids. In other words, in the NaCl structure, each cation is surrounded by four anions in the (100) plane and two ions in the perpendicular plane, which are equidistant from the cation. As a result, octahedral coordination takes place. This is equally true for anions. Therefore, the ratio of the coordination numbers of the sublattices is 6:6.
Structure of cesium chloride CsCl (double PC lattice),
shown in Fig. 1.13, b, consists of two primitive cubic lattices, shifted by half the volumetric diagonal. The fact is that cesium ions are larger than sodium ions and cannot fit into the octahedral (and even more so in the tetrahedral) voids of the chlorine lattice, if it were of the fcc type, as in the NaCl structure. In the CsCl structure, each cesium ion is surrounded by eight chlorine ions and vice versa.
Other halides also crystallize into structures of this type, for example Cs (Br, I), Rb (Br, I), Tl (Br, Cl), semiconductor compounds of the AIV BVI type and many alloys of rare earth elements. Similar structures are also observed in heteropolar ionic compounds.
1.4.3. Semiconductor crystals
Diamond structure is a combination of two face-centered gratings inserted into one another and shifted along the spatial diagonal by a quarter of the length (Fig. 1.14, a). Each atom is surrounded by four, which are located at the vertices of the tetrahedron (thick lines in Fig. 1.14, a). All bonds in the diamond structure are equal, directed along<111>and make angles of 109º 28" with each other. The diamond lattice belongs to loosely packed structures with coordination number z = 4. In the structure of diamond, germanium, silicon, and gray tin crystallize. In addition to diamond, elementary semiconductors - silicon Si, germanium Ge - also crystallize in this type of structure , gray tin Sn.
Sphalerite structure(double fcc lattice). If two auxiliary face-centered cubic lattices are formed by different atoms, then a new structure arises, called the ZnS sphalerite structure or zinc blende(Fig. 1.14, b).

Chapter 1. Elements of crystal physics
Rice. 1 .14. Structures of diamond (a), phalerite (b), wurtzite (c). Tetrahedral bonds are highlighted in bold lines.
Many semiconductor compounds of type AIII BV (gallium arsenide GaA s, gallium phosphide GaP, indium phosphide InP, indium antimonide I nSb, etc.) and type AII BVI (zinc selenide ZnSe, zinc telluride ZnTe, cadmium sulfide CdS, selenide) have this structure. cadmium
The structure of sphalerite is identical to the structure of diamond with a tetrahedral environment of atoms (Fig. 1.14, a), only one fcc sublattice is occupied by gallium Ga atoms, and the other by arsenic As atoms. There is no center of symmetry in a GaAs cell, i.e. the structure is polar in four directions.< 111 >. A difference is observed between the close packed 111) and (111) planes: if one of them contains Ga atoms, the other contains As atoms. This causes anisotropy in surface properties (microhardness, adsorption, chemical etching, etc.).
In the sphalerite structure, the triangular bases of the tetrahedrons of any layer are oriented in the same way as the bases of the tetrahedrons of the previous layer.
Wurtzite structure(with a dual GPU lattice), shown in Fig. 1.14, c, is characteristic of the hexagonal modification of zinc sulfide. Semiconductors close to ZnS, for example, cadmium sulfide CdS and cadmium selenide CdSe, have this structure. Most AII–VI compounds are characterized by a “sphalerite–wurtzite” phase transition. The wurtzite structure is realized if the nonmetal atom has small sizes and high electronegativity.
In Fig. Figure 1.14c shows a primitive wurtzite cell for ZnS in the form of a straight prism with a rhombus at the base and an angle of 120° at the center of a hexagon formed by three such prisms (two of which are shown in the figure).
CRYSTAL STRUCTURE
CRYSTAL STRUCTURE
The arrangement of atoms, ions, molecules in a crystal. Crystal with definition chem. f-loy has an inherent kinetic system, which has three-dimensional periodicity - crystal lattice. The term "K.s." are used instead of the term “crystal lattice” when talking about lattice energy, lattice dynamics, and the lattice as a specific structure of a particular chemical. compounds, about the description of the atomic structure of specific compounds and their modifications. Geom. description of a specific K. s. consists of indicating the coordinates of the centers of atoms in the unit cell of a crystal, which makes it possible to determine interatomic distances and thereby study the geome. features of K. s.
Basic methods of research K. s. are diffraction - X-ray structural analysis, neutron diffraction, electron diffraction. Diffraction methods provide continuous scattering matter averaged over time and over the entire volume of a crystal in a unit cell. X-ray methods analysis, the electron density in the crystal is obtained, which is calculated as a Fourier series:
Where x, y, z- coordinates in the unit cell, - its volume, F hkl- coefficient Fourier, called structural amplitudes. The electron density distribution in a cell can be approximately represented as the sum of the electron densities of atoms ( r i):

Where i- time-averaged, i.e., smeared by thermal motion, distributions of electrons in an atom. Maxima (1) correspond to atoms - electron density clumps, which makes it possible to find the coordinates of their centers r (x, y, z) and create a geom. model, establishing interatomic distances with an accuracy of 0.0001 - 0.00001 nm.
In neutronography, similar to (1) for the amplitudes of nuclear scattering of a crystal F neuters determine the distribution of nuclear density n(r)cells, i.e., a probabilistic distribution of nuclei smeared by thermal motion (see. Structural neutron diffraction). If the atoms have a magnetic moment is determined by neutron diffraction magnetically. K.s. - spin density distribution (see. Magnetic). In electron diffraction by amplitudes F el according to (1) determine the distribution of electrostatic. (total - nuclei and electrons) potential ( r). The position of the maxima of all three distributions coincides - this is the time-average position of the centers of atoms (nuclei) in the unit cell.
Geometric model. To create a geom. models K. s. necessary: knowledge of the parameters of the unit cell (repetition parallelepiped) of the structure - in the general case, the lengths of its edges a, b, c and angles; an indication of the symmetry of a space system, that is, whether it belongs to one of 230 space groups (see Crystal symmetry) and thus - like Bravais lattice;.
indication of the coordinates of all chemically different atoms and the symmetry of their positions. To do this, it is enough to know the coordinates of the atoms in the symmetrically independent part of the cell, from which it is possible, taking into account the operations of the space group, to deduce the position of all the atoms of the cosmos. On this basis, interatomic distances, mutual coordination of atoms, etc. are calculated. characteristics of K. s. Graphically K. s. depicted by the arrangement of atoms (“balls”) in a unit cell (Fig. 1, a). If necessary, large touching “balls” can show the contacts of atoms in close-packed inorganic materials. (Fig. 1, b) or molecular structures. For the image of ionic K. s. Polyhedra are often used, with anions at the vertices and cations at the centers (Fig. 1c).
Geom. analysis of K. s. made it possible to develop a number of generalizations and laws of the atomic structure of crystals - ideas about atomic radii, about types of chemicals. bonds in crystals (ionic, covalent, metallic, van der Waals, hydrogen), rules for the closest packing of atoms and molecules in crystals, bonds of crystals. with the properties of crystals (see Crystal chemistry). Analysis of K. s. and its symmetry serves as the starting point for energy calculations. spectrum, interpretation of physical properties of the crystal (see Crystal physics).
Parameters of unit cells of some crystals
| Types of crystals | Unit cell periods, nm | Number of atoms in a unit cell | ||
| Inorganic and simple molecular compounds | up to hundreds | |||
| Complex organic compounds | up to thousands | |||
| Viruses | ||||
More than 100 thousand K. s. have been studied. diff. substances, of which approx. 20 thousand inorganic K. s. elements, diff. compounds, minerals, the rest - most - are organic. K. s. Lattice periods vary. crystals range from fractions to hundreds of nm (table). X-ray structural analysis of K. s. organic connections there are max. accurate and reliable method for determining spatial and chemical the structure of their constituent molecules. Several have been studied. hundreds K. s. the most complex substances in biol. origin: proteins, nucleic acids, viruses (see. Biological crystal). There are international computer data banks that describe everything inorganic, organic. and biological K. s.

Rice. 1. Models of crystal structures: A- diamond, b - NaCl chloride, V- bafertisite BaFe 2 Tl (Si 2 O 7)O(OH).
Modern Precision diffraction methods make it possible, in addition to the coordinates of atoms (geom. models), to determine other characteristics of cosmic structures.
It is possible experimentally to determine in detail the anharmonicity of the thermal vibrations of the atoms of a cosmic system, which is described by tensors of a higher rank. The surface characterizing the vibrations is no longer a triaxial Gaussian ellipsoid and does not have a center of symmetry. Anharmonicity parameters make it possible to relate the nature of atomic vibrations to acoustic, ferroelectric. properties of crystals, indicate possible displacements of atoms during phase transitions in high-temperature modifications of crystals. Frequencies of atomic vibrations in cosmic rays. are about 10 12 Hz, they are determined spectroscopically. methods, method inelastic neutron scattering(cm. Vibrations of the crystal lattice).

Rice. 2. Ellipsoids of thermal vibrations of atoms in the lattice: A- general case of arbitrary orientation; b- anisotropy of vibrations in the structure, - acetylene - nickel bis-cyclopentadiene at 300 K. On the left - acetylene, on the right - cyclopentadiene.
Subtracting from the observed distribution ( r)(1) distribution ( r)(2), one can find the deformation electronic crystal structure.
The fact is that expression (2) is the sum of free “pro-atoms” of the crystal, smeared by thermal movement, the electron density of which is not changed due to the formation of chemicals. bonds in the crystal system, and expression (1) corresponds to the electron density of the crystal in which all these bonds were formed. Despite the fact that the values are small, they make it possible to identify a number of subtle details of the system. (Fig. 3). Thus, the appearance of a maximum in the place of a “proatom” indicates an excess of electrons in it, i.e., that this one is negatively charged, since it is an anion, and the appearance of a minimum indicates that it is a cation; The degree of ionization can be estimated from the dr def value. In ionic K. s. electrons are redistributed between atoms, but in the “interatomic space” practically = 0. In metallic K. s. Some of the electrons of the atoms are shared and form a uniform electron density in the interatomic space. In covalent crystals, it clearly reveals peaks between atoms, corresponding to pairs of electrons forming a covalent bond. In molecular crystals, peaks corresponding to lone electron pairs of atoms are recorded (Fig. 3). Math. treatment ( r) and allows you to find the distribution of electrostatic. electron potential, potential energy, field gradient on atoms, etc. Using electron diffraction, you can find the total (nuclei and electrons) deformation. atoms and identify them.

Rice. 3. Deformation electron density of cyanuric acid. The peaks on the bonds are valence bonding electrons, near the O atom - a lone electron pair.
Defects. C.s., in which all positions are filled with atoms, is called. ideal K. s. However, in reality K. s. has a number of defects - point (displacement of atoms from ideal positions, replacement of these atoms by impurity atoms, vacancies, interstitial atoms, etc.), linear and two-dimensional (dislocations, errors in the application of layers, etc.) (see. Defects in crystals). If the number of point defects is large, it is possible to record the average change over all cells in the electronic density of the CS, for example. in ruby A1 2 0 3 +0.05% Cr, where Cr replaces the positions of A1. In the structures of subtraction or interstitial solid solutions, analysis of br provides information about the occupancy of certain positions by atoms.
Along with crystalline substances in which atoms vibrate around a fixed point. equilibrium positions, there are crystals, in which the dep. atoms, their groups or whole molecules statistically occupy different. provisions (see solid). Thermal molecules in certain crystals. structures is such that while maintaining the position of the center of gravity, they can be in a spherical state. or cylindrical rotation. In some crystals, in the presence of a rigid three-dimensional periodicity. framework of a structure consisting of only atoms, certain ions can freely migrate and flow through the channels of the framework (see. Ionic superconductors). The migration paths of charged ions are fixed by the distribution dr def. Similarly in frame frame systems, for example. zeolites, inside the voids there may be organic molecules. substances also recorded by dr.

Rice. 4. Electron microscopic image of the atomic structure of a phosphorus cluster in silicon.
The specific location of defects in a real code. It is also studied using X-ray and neutron topography, electron microscopy (Fig. 4), etc.
Complex K. s. Along with ideal three-dimensional periodic K. s. there are other types of crystalline. ordering of atoms. Thus, in superstructures against the “background” of a regular three-dimensional lattice, complementarity is observed. orderliness with periods that are multiples of one or two periods of an ideal cosmic system, due, for example, to the distribution of magnetic fields. moments of atoms, electrical dipoles, etc. Sometimes the period of such a superstructure is not a multiple of the period of the main lattice, and then the Q.s. called disproportionate. K. s. with periodic ones in k.-l. direction of inclusions of foreign atoms is called. modulated. Heterostructures artificially prepared in microelectronics, for example. AlAs-GaAs, have a common, the same crystalline. lattice (in the sense of equality of periods), but they alternate layers of one or another composition (Fig. 5). There are K. s. (for example, layered silicates) with a disordered overlay of two or more types of fixed layers. buildings, for example structures of articulated “ribbons” or “columns” of fixed composition. All this is actually a coherent coupling in a single crystal at the atomic level of micro-sections of decomposition. K. s.

Rice. 5. Electron micrograph of the arrangement of atoms in the AlAs-GaAs heterostructure (magnification 10 6).
More complex disturbances of order, leading to partial or complete loss of fundamentals. sign of K. s. - long-range order (see. Long-range and short-range order), observed in the structure of polymers, liquid crystals, quasicrystals.
K. s. specific substances are classified according to symmetry and chemical type. connections. Many substances of different chemicals. composition, but with the same ratio of the number of atoms, they have geometrically similar K.s., which is called. from structure (eg MgO and TiN - structural type of NaCl). From the symmetry of K. s. it is possible to predict the possible physical factors in a given crystal. properties. Quantitative characteristics of various properties, for example, elastic, optical, electrical, etc., can be linked to the specific arrangement of atoms in a cosmic structure, and sometimes can be directly calculated from the cosmic structure. (cm. Crystals),
Lit.: Structure reports. Publ. for the Intern. Union of Crystallography, Utrecht, 1951-87 - ; Molecular structures and dimensions. Bibliography, ed. by O. Kennard and D. Watson, v. 1-15, Utrecht, 1971-84; Modern, vol. 2, M., 1979; Neutrons and solids, vol. 2, M., 1981; Vainshtein B.K., Structural classification of states of matter, in the book: Crystallography and crystal chemistry, M., 1986; Walls A., Structural Inorganic Chemistry, trans. from English, vol. 1, M., 1987. B. K. Weinstein.
Physical encyclopedia. In 5 volumes. - M.: Soviet Encyclopedia. Editor-in-chief A. M. Prokhorov. 1988 .
Send your good work in the knowledge base is simple. Use the form below
Students, graduate students, young scientists who use the knowledge base in their studies and work will be very grateful to you.
Crystals (from the Greek xeufbllpt, originally - ice, later - rock crystal, crystal) are solid bodies in which atoms are arranged regularly, forming a three-dimensional periodic spatial arrangement - a crystal lattice.
Crystals are solid substances that have a natural external shape of regular symmetrical polyhedra based on their internal structure, that is, on one of several specific regular arrangements of particles (atoms, molecules, ions) that make up the substance.
Properties:
Uniformity. This property is manifested in the fact that two identical elementary volumes of a crystalline substance, identically oriented in space, but cut out at different points of this substance, are absolutely identical in all their properties: they have the same color, specific gravity, hardness, thermal conductivity, electrical conductivity and etc.
It must be borne in mind that real crystalline substances very often contain permanent impurities and inclusions that distort their crystal lattices. Therefore, absolute homogeneity in real crystals often does not exist.
Anisotropy of crystals
Many crystals have the property of anisotropy, that is, the dependence of their properties on direction, while in isotropic substances (most gases, liquids, amorphous solids) or pseudoisotropic (polycrystals) properties do not depend on directions. The process of inelastic deformation of crystals always occurs along well-defined slip systems, that is, only along certain crystallographic planes and only in a certain crystallographic direction. Due to the inhomogeneous and unequal development of deformation in different areas of the crystalline medium, intense interaction occurs between these areas through the evolution of microstress fields.
At the same time, there are crystals in which anisotropy is absent.
In the physics of martensitic inelasticity, rich experimental material has been accumulated, especially on the issues of shape memory effects and transformation plasticity. The most important position of crystal physics about the predominant development of inelastic deformations almost exclusively through martensitic reactions has been experimentally proven. But the principles of constructing a physical theory of martensitic inelasticity are unclear. A similar situation occurs in the case of crystal deformation by mechanical twinning.
Significant progress has been made in the study of dislocation plasticity of metals. Here, not only the basic structural and physical mechanisms of the implementation of inelastic deformation processes are understood, but also effective methods for calculating the phenomena have been created.
The ability to self-distill is the property of crystals to form faces during free growth. So. If a ball carved from some substance, for example table salt, is placed in its supersaturated solution, then after some time this ball will take the shape of a cube. In contrast, a glass bead will not change its shape since an amorphous substance cannot self-distill.
Constant melting point. If you heat a crystalline body, its temperature will increase to a certain limit; with further heating, the substance will begin to melt, and the temperature will remain constant for some time, since all the heat will go to destroy the crystal lattice. The temperature at which melting begins is called the melting point.
Crystal taxonomy
Crystal structure
The crystal structure, being individual for each substance, refers to the basic physical and chemical properties of this substance. A crystalline structure is a collection of atoms in which a certain group of atoms, called a motivic unit, is associated with each point of the crystal lattice, and all such groups are identical in composition, structure and orientation relative to the lattice. It can be considered that the structure arises as a result of the synthesis of the lattice and the motif unit, as a result of the propagation of the motif unit by the translation group.
In the simplest case, the motif unit consists of a single atom, for example in copper or iron crystals. The structure that arises on the basis of such a motif unit is geometrically very similar to a lattice, but still differs in that it is composed of atoms rather than points. This circumstance is often not taken into account, and the terms “crystal lattice” and “crystal structure” for such crystals are used as synonyms, which is not strict. In cases where the motif unit is more complex in composition - consists of two or more atoms, there is no geometric similarity of the lattice and structure, and the displacement of these concepts leads to errors. For example, the structure of magnesium or diamond does not coincide geometrically with the lattice: in these structures, the motif units consist of two atoms.
The main parameters characterizing the crystal structure, some of which are interrelated, are the following:
§ type of crystal lattice (systemony, Bravais lattice);
§ number of formula units per unit cell;
§ space group;
§ unit cell parameters (linear dimensions and angles);
§ coordinates of atoms in a cell;
§ coordination numbers of all atoms.
Structural type
Crystal structures that have the same space group and the same arrangement of atoms in crystal chemical positions (orbits) are combined into structural types.
The best known structural types are copper, magnesium, b-iron, diamond (simple substances), sodium chloride, sphalerite, wurtzite, cesium chloride, fluorite (binary compounds), perovskite, spinel (ternary compounds).
Crystal cell
The constituent particles of this solid form a crystal lattice. If crystal lattices are stereometrically (spatially) identical or similar (have the same symmetry), then the geometric difference between them lies, in particular, in different distances between the particles occupying lattice sites. The distances between particles themselves are called lattice parameters. The lattice parameters, as well as the angles of geometric polyhedra, are determined by physical methods of structural analysis, for example, methods of X-ray structural analysis.
Posted on http://www.allbest.ru/
Rice. Crystal cell
Often solids form (depending on conditions) more than one form of crystal lattice; such forms are called polymorphic modifications. For example, among simple substances, rhombic and monoclinic sulfur, graphite and diamond are known, which are hexagonal and cubic modifications of carbon; among complex substances, quartz, tridymite and cristobalite are various modifications of silicon dioxide.
Types of crystals
It is necessary to separate the ideal and real crystal.
Perfect Crystal
It is, in fact, a mathematical object that has complete, inherent symmetry, idealized smooth smooth edges.
Real crystal
It always contains various defects in the internal structure of the lattice, distortions and irregularities on the faces and has a reduced symmetry of the polyhedron due to the specific growth conditions, heterogeneity of the feeding medium, damage and deformations. A real crystal does not necessarily have crystallographic faces and a regular shape, but it retains its main property - the regular position of atoms in the crystal lattice.
Defects in the crystal lattice (real structure of crystals)
In real crystals there are always deviations from the ideal order in the arrangement of atoms, called imperfections or lattice defects. Based on the geometry of the lattice disruptions they cause, defects are divided into point, linear and surface.
Point defects
In Fig. Figure 1.2.5 shows different types of point defects. These are vacancies - empty lattice sites, “own” atoms in interstices, and impurity atoms in lattice sites and interstices. The main reason for the formation of the first two types of defects is the movement of atoms, the intensity of which increases with increasing temperature.
Rice. 1.2.5. Types of point defects in the crystal lattice: 1 - vacancy, 2 - atom in an interstitial site, 3 and 4 - impurity atoms in a site and interstitial site, respectively
Around any point defect, a local distortion of the lattice with a radius R of 1...2 lattice periods occurs (see Fig. 1.2.6), therefore, if there are many such defects, they affect the nature of the distribution of interatomic bond forces and, accordingly, the properties of the crystals.
Rice. 1.2.6. Local distortion of the crystal lattice around a vacancy (a) and an impurity atom at a lattice site (b)
Linear defects
Linear defects are called dislocations. Their appearance is caused by the presence in certain parts of the crystal of “extra” atomic half-planes (extraplanes). They arise during the crystallization of metals (due to a violation of the order of filling atomic layers) or as a result of their plastic deformation, as shown in Fig. 1.2.7.
Rice. 1.2.7. Formation of an edge dislocation () as a result of partial displacement of the upper part of the crystal under the influence of force: ABCD - slip plane; EFGН - extraplane; EN - edge dislocation line
It can be seen that, under the influence of a shear force, a partial shift of the upper part of the crystal occurred along a certain sliding plane (“light shear”) ABCD. As a result, the extraplane EFGH was formed. Since it does not continue downwards, an elastic distortion of the lattice appears around its edge EH with a radius of several interatomic distances (i.e. 10 -7 cm - see topic 1.2.1), the extent of this distortion is many times greater (can reach up to 0.1...1 cm).
This imperfection of the crystal around the edge of the extraplane is a linear lattice defect and is called an edge dislocation.
The most important mechanical properties of metals - strength and ductility (see topic 1.1) - are determined by the presence of dislocations and their behavior when the body is loaded.
Let us dwell on two features of the mechanism of dislocation movement.
1. Dislocations can very easily (at a low load) move along the slip plane through the “relay race” movement of the extraplane. In Fig. Figure 1.2.8 shows the initial stage of such a movement (a two-dimensional pattern in a plane perpendicular to the edge dislocation line).
Rice. 1.2.8. The initial stage of the relay movement of an edge dislocation (). A-A - sliding plane, 1-1 extraplane (initial position)
Under the influence of force, the atoms of the extraplane (1-1) tear off the atoms (2-2) located above the sliding plane from the plane (2-3). As a result, these atoms form a new extraplane (2-2); the atoms of the “old” extraplane (1-1) occupy the vacant spaces, completing the construction of the plane (1-1-3). This act means the disappearance of the “old” dislocation associated with the extra-plane (1-1), and the emergence of a “new” one associated with the extra-plane (2-2), or, in other words, the transfer of the “relay baton” - the dislocation to one interplanar distance. This relay movement of the dislocation will continue until it reaches the edge of the crystal, which will mean a shift of its upper part by one interplanar distance (i.e., plastic deformation).
This mechanism does not require much effort, because consists of successive microdisplacements affecting only a limited number of atoms surrounding the extraplane.
2. It is obvious, however, that such ease of sliding of dislocations will be observed only in the case when there are no obstacles in their path. Such obstacles are any lattice defects (especially linear and surface!), as well as particles of other phases, if they are present in the material. These obstacles create lattice distortions, overcoming which requires additional external forces, and therefore can block the movement of dislocations, i.e. make them motionless.
Surface defects
All industrial metals (alloys) are polycrystalline materials, i.e. consist of a huge number of small (usually 10 -2 ... 10 -3 cm), chaotically oriented crystals, called grains. It is obvious that the lattice periodicity inherent in each grain (single crystal) is disrupted in such a material, since the crystallographic planes of the grains are rotated relative to each other by an angle b (see Fig. 1.2.9), the value of which varies from fractions to several tens of degrees.
Rice. 1.2.9. Scheme of the structure of grain boundaries in a polycrystalline material
The boundary between grains is a transition layer up to 10 interatomic distances wide, usually with a disordered arrangement of atoms. This is a place where dislocations, vacancies, and impurity atoms accumulate. Therefore, in the bulk of a polycrystalline material, grain boundaries are two-dimensional, surface defects.
The influence of lattice defects on the mechanical properties of crystals. Ways to increase the strength of metals.
Strength is the ability of a material to resist deformation and destruction under the influence of external load.
The strength of crystalline bodies is understood as their resistance to an applied load, tending to move or, in the limit, tear off one part of the crystal relative to another.
The presence of mobile dislocations in metals (already during the crystallization process up to 10 6 ... 10 8 dislocations appear in a cross section equal to 1 cm 2) leads to their reduced resistance to loading, i.e. high ductility and low strength.
Obviously, the most effective way to increase strength is to remove dislocations from the metal. However, this path is not technologically advanced, because dislocation-free metals can only be obtained in the form of thin threads (so-called “whiskers”) with a diameter of several microns and a length of up to 10 microns.
Therefore, practical methods of strengthening are based on braking, blocking mobile dislocations by sharply increasing the number of lattice defects (primarily linear and surface!), as well as creating multiphase materials
Such traditional methods of increasing the strength of metals are:
– plastic deformation (the phenomenon of work hardening or hardening),
– thermal (and chemical-thermal) treatment,
– alloying (introduction of special impurities) and, the most common approach, is the creation of alloys.
In conclusion, it should be noted that an increase in strength based on blocking mobile dislocations leads to a decrease in ductility and impact strength and, accordingly, the operational reliability of the material.
Therefore, the question of the degree of hardening must be decided individually, based on the purpose and operating conditions of the product.
Polymorphism in the literal sense of the word means multiformity, i.e. a phenomenon when substances of the same chemical composition crystallize in different structures and form crystals of different syngogies. For example, diamond and graphite have the same chemical composition, but different structures; both minerals differ sharply in physical properties. properties. Another example is calcite and aragonite - they have the same composition of CaCO 3, but represent different polymorphs.
The phenomenon of polymorphism is associated with the conditions of formation of crystalline substances and is due to the fact that only certain structures are stable under various thermodynamic conditions. Thus, metal tin (the so-called white tin) when the temperature drops below -18 C 0 becomes unstable and crumbles, forming “gray tin” of a different structure
Isomorphism. Metal alloys are crystalline structures of variable composition, in which atoms of one element are located in the interstices of the crystal lattice of another. These are the so-called solid solutions of the second kind.
Unlike solid solutions of the second kind, in solid solutions of the first kind, atoms or ions of one crystalline substance can be replaced by atoms or ions of another. The latter are located at the nodes of the crystal lattice. Solutions of this kind are called isomorphic mixtures.
Conditions necessary for the manifestation of isomorphism:
1) Only ions of the same sign can be replaced, i.e., cation by cation, and anion by anion
2) Only atoms or ions of similar size can be replaced, i.e. the difference in the ionic radii should not exceed 15% for perfect isomorphism and 25% for imperfect isomorphism (for example, Ca 2+ on Mg 2+)
3) Only ions that are close in degree of polarization (i.e. in the degree of ionicity-covalence of the bond) can be replaced.
4) Only elements that have the same coordination number in a given crystal structure can be replaced
5) isomorphic substitutions should occur in this way. So that the electrostatic balance of the crystal lattice is not disturbed.
6) isomorphic substitutions occur in the direction of increasing lattice energy.
Types of isomorphism. There are 4 types of isomorphism:
1) isovalent isomorphism is characterized by the fact that in this case there are ions of the same valence and the difference in the sizes of ionic radii should not be more than 15%
2) heterovalent isomorphism. In this case, replacement of ions of different valence occurs. With such a substitution, one ion cannot be replaced by another without disturbing the electrostatic balance of the crystal lattice, therefore, with heterovalent isomorphism, not an ion is replaced, as with heterovalent isomorphism, but a group of ions of a certain valency is replaced by another group of ions while maintaining the same total valence.
In this case, it is necessary to always remember that the replacement of an ion of one valency with an ion of another is always associated with compensation of valency. This compensation can occur in both the cationic and anionic parts of the compounds. In this case, the following conditions must be met:
A) the sum of the valences of the replaced ions must be equal to the sum of the valences of the replacing ions.
B) the sum of the ionic radii of the replaced ions should be close to the sum of the ionic radii of the replacing ions and may differ from it by no more than 15% (for perfect isomorphism)
3) isostructural. What occurs is not one ion being replaced by another or a group of ions by another group, but an entire “block” of one crystal lattice is being replaced by another similar “block”. This can only happen if the structures of the minerals are of the same type and have similar unit cell sizes.
4) isomorphism of a special kind.
crystal lattice defect dislocation
Posted on Allbest.ru
Similar documents
Characteristics of the piezoelectric effect. Study of the crystal structure of the effect: model consideration, crystal deformations. Physical mechanism of the inverse piezoelectric effect. Properties of piezoelectric crystals. Applying an effect.
course work, added 12/09/2010
Information about vibrations of crystal lattices, functions describing their physical quantities. Crystallographic coordinate systems. Calculation of the interaction energy of atoms in covalent crystals, the vibration spectrum of the crystal lattice of barium tungstate.
thesis, added 01/09/2014
Passage of current through electrolytes. Physical nature of electrical conductivity. The influence of impurities and crystal structure defects on the resistivity of metals. Resistance of thin metal films. Contact phenomena and thermoelectromotive force.
abstract, added 08/29/2010
Concept and classification of defects in crystals: energy, electronic and atomic. The main imperfections of crystals, the formation of point defects, their concentration and the speed of movement along the crystal. Particle diffusion due to vacancy movements.
abstract, added 01/19/2011
The essence of polymorphism, the history of its discovery. Physical and chemical properties of polymorphic modifications of carbon: diamond and graphite, their comparative analysis. Polymorphic transformations of liquid crystals, thin films of tin diiodide, metals and alloys.
course work, added 04/12/2012
Crystalline and amorphous states of solids, causes of point and linear defects. Nucleation and growth of crystals. Artificial production of precious stones, solid solutions and liquid crystals. Optical properties of cholesteric liquid crystals.
abstract, added 04/26/2010
History of the development of the concept of liquid crystals. Liquid crystals, their types and basic properties. Optical activity of liquid crystals and their structural properties. Fredericks effect. The physical principle of operation of LCD devices. Optical microphone.
tutorial, added 12/14/2010
Crystallization is the process of transition of a metal from a liquid to a solid state with the formation of a crystalline structure. Scheme of seam formation during arc welding. Key factors and conditions necessary for the growth of liquid metal crystals to begin.
presentation, added 04/26/2015
Study of the structure (formation by crystallites arranged in a chaotic manner) and methods of production (melt cooling, vapor deposition, bombardment of crystals with neurons) of glasses. Familiarization with the processes of crystallization and glass transition.
abstract, added 05/18/2010
Defects in real crystals, the operating principle of bipolar transistors. Distortion of the crystal lattice in interstitial and substitutional solid solutions. Surface phenomena in semiconductors. Transistor parameters and emitter current transfer coefficient.
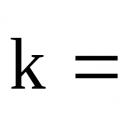 Reaction rate constant Guidelines for laboratory work
Reaction rate constant Guidelines for laboratory work Is there life after death?
Is there life after death? Scientists: Earth is a “space prison” for humanity scientists: Earth is a space prison in the universe
Scientists: Earth is a “space prison” for humanity scientists: Earth is a space prison in the universe